

Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway
- PMID: 16365149
- PMCID: PMC2212966
- DOI: 10.1084/jem.20051047
Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway
Abstract
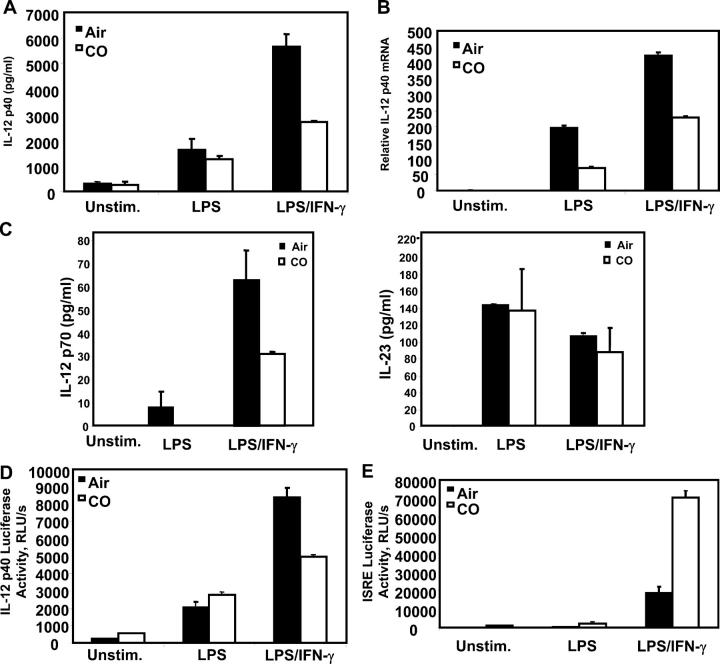
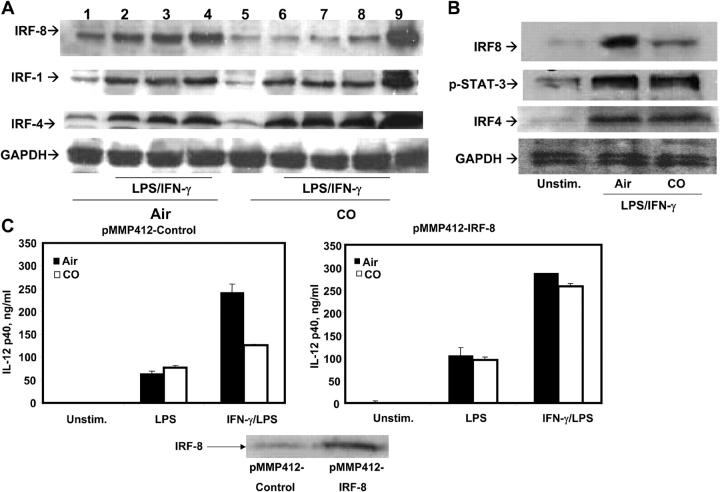
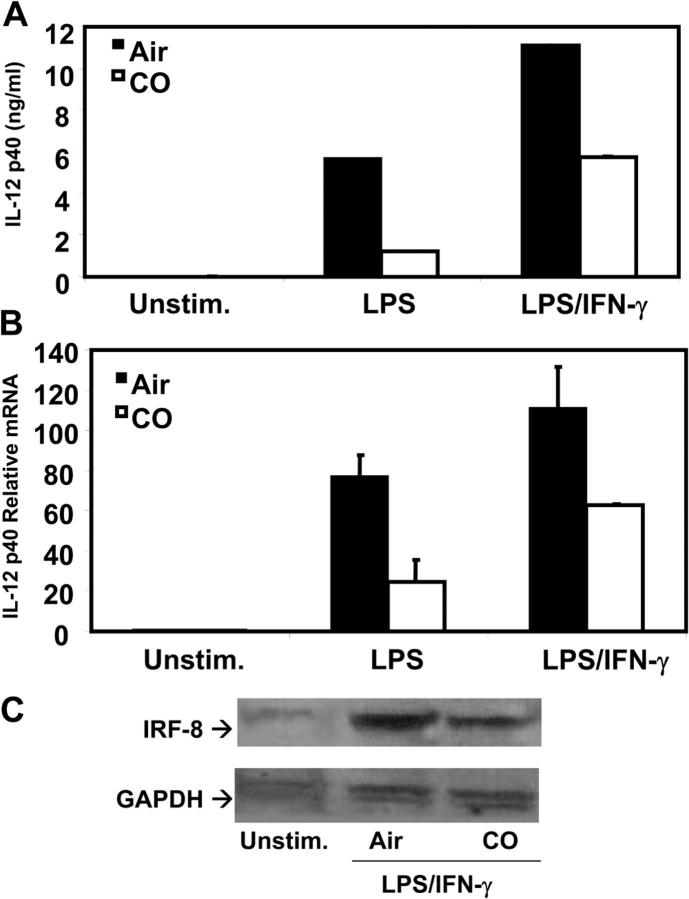
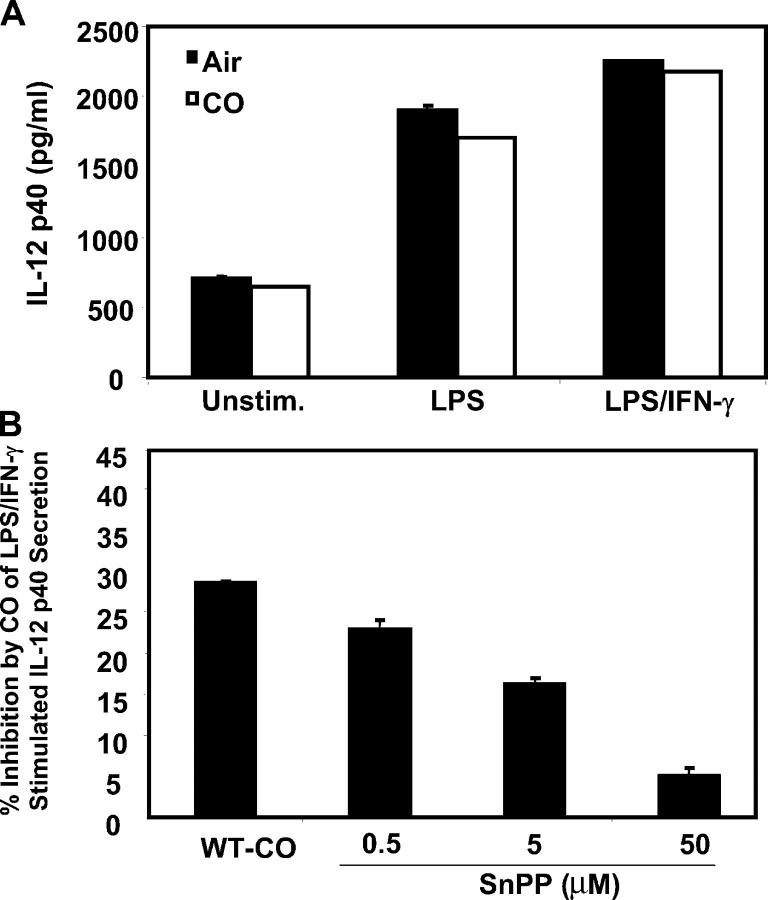
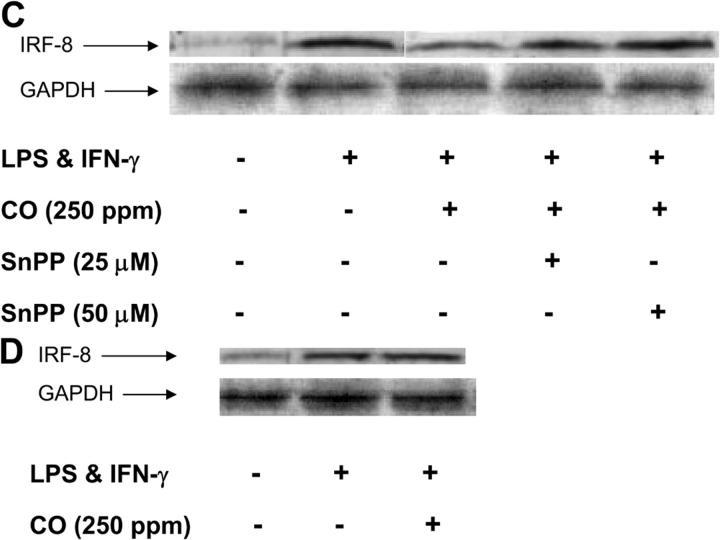
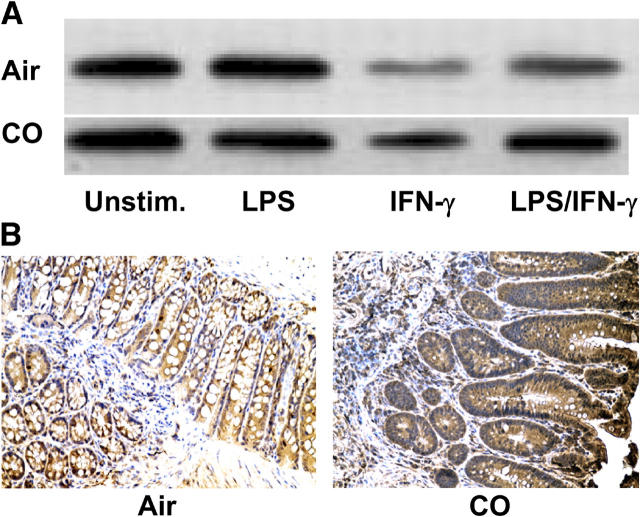
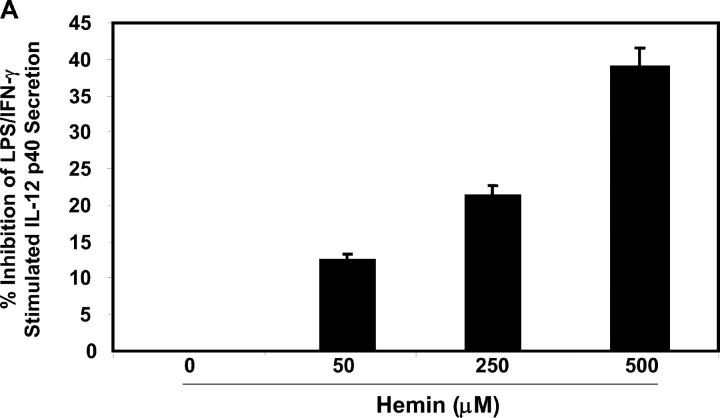
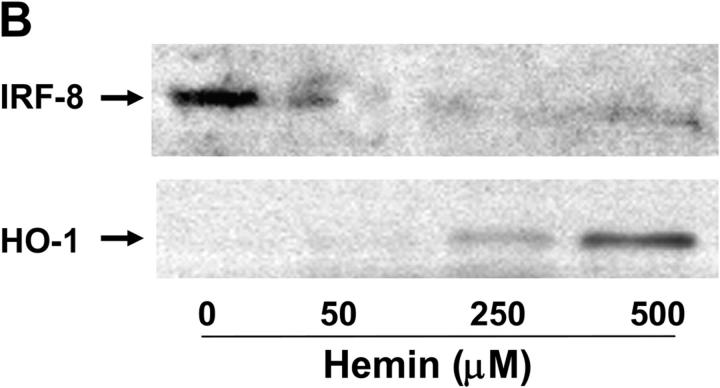
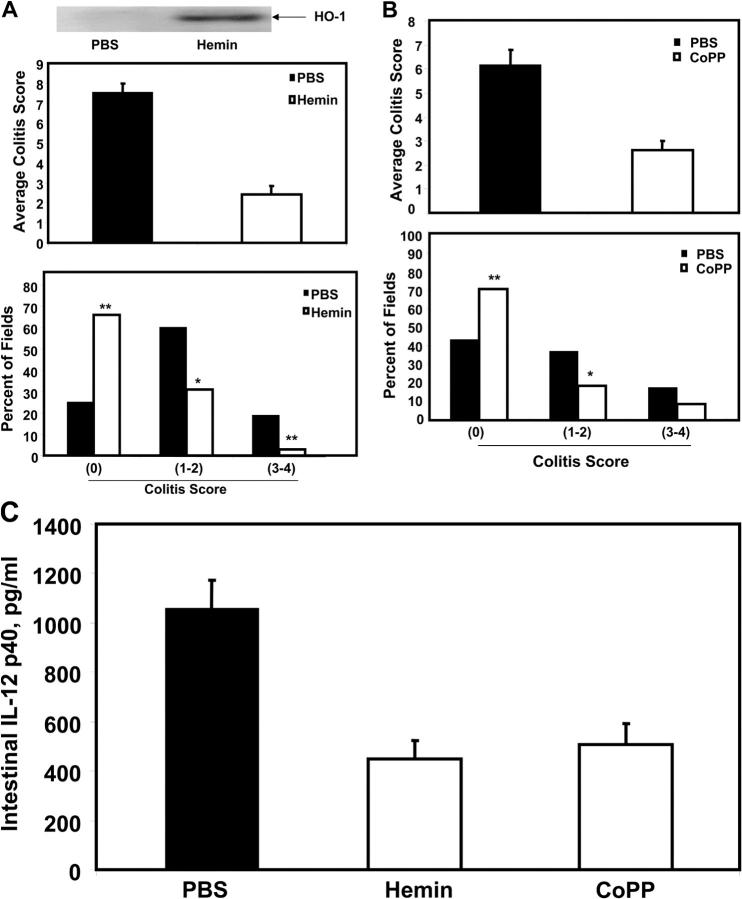
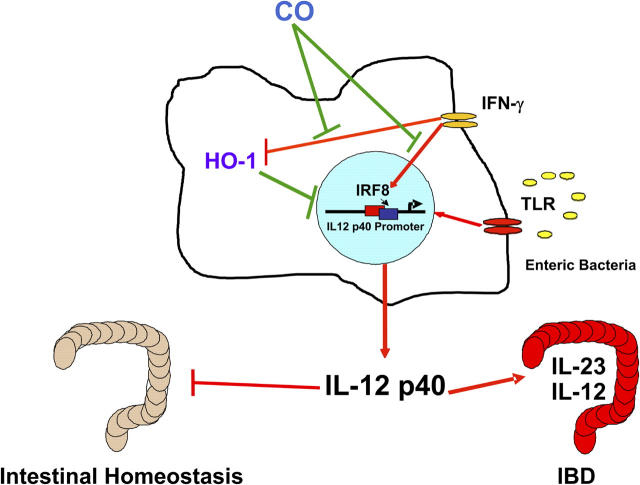
Heme oxygenase (HO)-1 and its metabolic product carbon monoxide (CO) play regulatory roles in acute inflammatory states. In this study, we demonstrate that CO administration is effective as a therapeutic modality in mice with established chronic colitis. CO administration ameliorates chronic intestinal inflammation in a T helper (Th)1-mediated model of murine colitis, interleukin (IL)-10-deficient (IL-10(-/-)) mice. In Th1-mediated inflammation, CO abrogates the synergistic effect of interferon (IFN)-gamma on lipopolysaccharide-induced IL-12 p40 in murine macrophages and alters IFN-gamma signaling by inhibiting a member of the IFN regulatory factor (IRF) family of transcription factors, IRF-8. A specific signaling pathway, not previously identified, is delineated that involves an obligatory role for HO-1 induction in the protection afforded by CO. Moreover, CO antagonizes the inhibitory effect of IFN-gamma on HO-1 expression in macrophages. In macrophages and in Th1-mediated colitis, pharmacologic induction of HO-1 recapitulates the immunosuppressive effects of CO. In conclusion, this study begins to elucidate potential etiologic and therapeutic implications of CO and the HO-1 pathway in chronic inflammatory bowel diseases.
Figures


Similar articles
-
An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis.J Immunol. 2011 May 1;186(9):5506-13. doi: 10.4049/jimmunol.1002433. Epub 2011 Mar 28. J Immunol. 2011. PMID: 21444764 Free PMC article.
-
Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine.Gastroenterology. 2003 Feb;124(2):377-91. doi: 10.1053/gast.2003.50060. Gastroenterology. 2003. PMID: 12557144
-
Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance.Gastroenterology. 2013 Apr;144(4):789-98. doi: 10.1053/j.gastro.2012.12.025. Epub 2012 Dec 22. Gastroenterology. 2013. PMID: 23266559 Free PMC article.
-
Heme oxygenase-1 and carbon monoxide in pulmonary medicine.Respir Res. 2003;4(1):7. doi: 10.1186/1465-9921-4-7. Epub 2003 Aug 7. Respir Res. 2003. PMID: 12964953 Free PMC article. Review.
-
Heme oxygenase-1/carbon monoxide: novel therapeutic strategies in critical care medicine.Curr Drug Targets. 2010 Dec;11(12):1485-94. doi: 10.2174/1389450111009011485. Curr Drug Targets. 2010. PMID: 20704552 Review.
Cited by
-
Disruption of monocyte-macrophage differentiation and trafficking by a heme analog during active inflammation.Mucosal Immunol. 2022 Feb;15(2):244-256. doi: 10.1038/s41385-021-00474-8. Epub 2021 Dec 16. Mucosal Immunol. 2022. PMID: 34916594 Free PMC article.
-
Review article: carbon monoxide in gastrointestinal physiology and its potential in therapeutics.Aliment Pharmacol Ther. 2013 Oct;38(7):689-702. doi: 10.1111/apt.12467. Epub 2013 Aug 28. Aliment Pharmacol Ther. 2013. PMID: 23992228 Free PMC article. Review.
-
Carbon Monoxide Being Hydrogen Sulfide and Nitric Oxide Molecular Sibling, as Endogenous and Exogenous Modulator of Oxidative Stress and Antioxidative Mechanisms in the Digestive System.Oxid Med Cell Longev. 2020 Apr 15;2020:5083876. doi: 10.1155/2020/5083876. eCollection 2020. Oxid Med Cell Longev. 2020. PMID: 32377300 Free PMC article. Review.
-
"CO in a pill": Towards oral delivery of carbon monoxide for therapeutic applications.J Control Release. 2021 Oct 10;338:593-609. doi: 10.1016/j.jconrel.2021.08.059. Epub 2021 Sep 2. J Control Release. 2021. PMID: 34481027 Free PMC article. Review.
-
Heme oxygenase-1 as a therapeutic target in inflammatory disorders of the gastrointestinal tract.World J Gastroenterol. 2010 Jul 7;16(25):3112-9. doi: 10.3748/wjg.v16.i25.3112. World J Gastroenterol. 2010. PMID: 20593496 Free PMC article. Review.
References
-
- Elson, C.O., R.B. Sartor, S.R. Targan, and W.J. Sandborn. 2003. Challenges in IBD research: updating the scientific agendas. Inflamm. Bowel Dis. 9:137–153. - PubMed
-
- Plevy, S., and L. Mayer. 2003. Meeting summary: signal transduction pathways in immune and inflammatory cells. November 30-December 3, 2000, Amelia Island, Florida, U.S.A. Inflamm. Bowel Dis. 9:28–33. - PubMed
-
- Jick, H., and A.M. Walker. 1983. Cigarette smoking and ulcerative colitis. N. Engl. J. Med. 308:261–263. - PubMed
-
- Boyko, E.J., T.D. Koepsell, D.R. Perera, and T.S. Inui. 1987. Risk of ulcerative colitis among former and current cigarette smokers. N. Engl. J. Med. 316:707–710. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
