

Genome-wide associations of gene expression variation in humans
- PMID: 16362079
- PMCID: PMC1315281
- DOI: 10.1371/journal.pgen.0010078
Genome-wide associations of gene expression variation in humans
Abstract
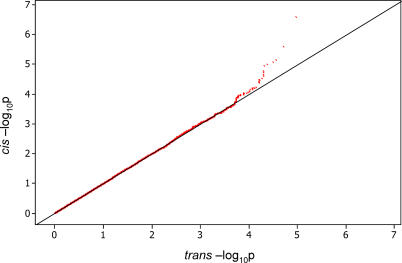
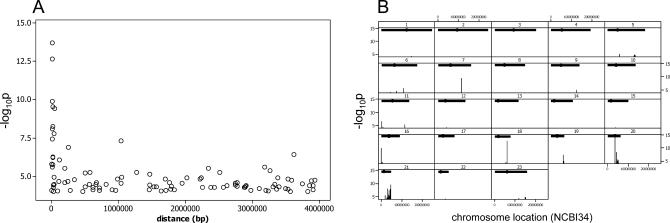
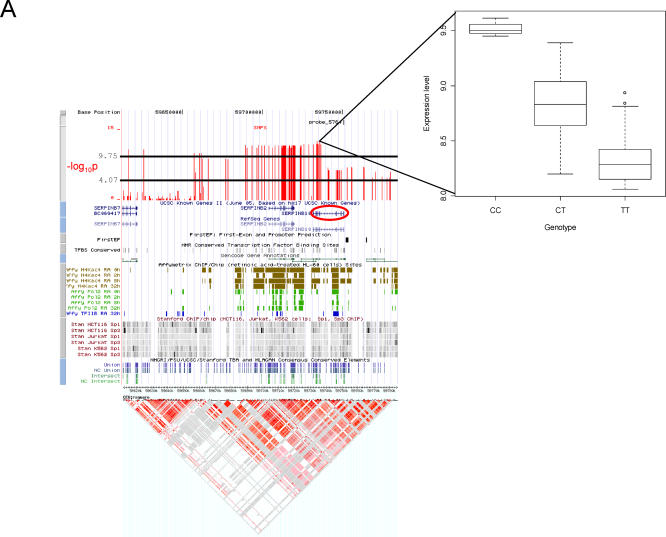
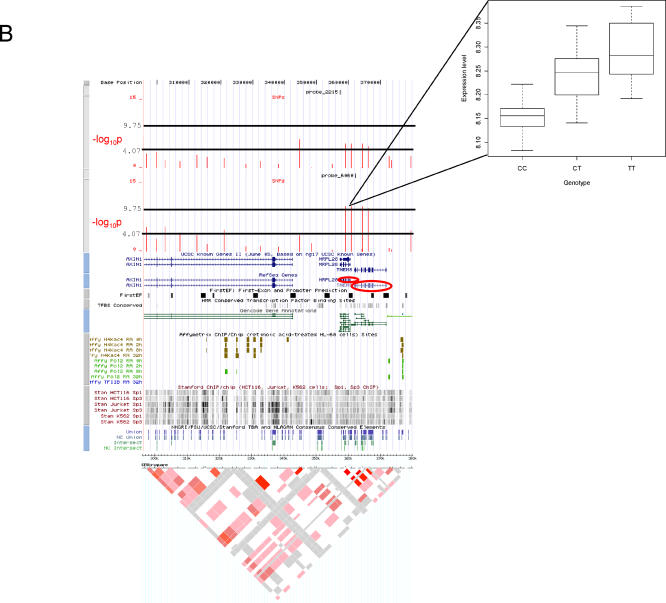
The exploration of quantitative variation in human populations has become one of the major priorities for medical genetics. The successful identification of variants that contribute to complex traits is highly dependent on reliable assays and genetic maps. We have performed a genome-wide quantitative trait analysis of 630 genes in 60 unrelated Utah residents with ancestry from Northern and Western Europe using the publicly available phase I data of the International HapMap project. The genes are located in regions of the human genome with elevated functional annotation and disease interest including the ENCODE regions spanning 1% of the genome, Chromosome 21 and Chromosome 20q12-13.2. We apply three different methods of multiple test correction, including Bonferroni, false discovery rate, and permutations. For the 374 expressed genes, we find many regions with statistically significant association of single nucleotide polymorphisms (SNPs) with expression variation in lymphoblastoid cell lines after correcting for multiple tests. Based on our analyses, the signal proximal (cis-) to the genes of interest is more abundant and more stable than distal and trans across statistical methodologies. Our results suggest that regulatory polymorphism is widespread in the human genome and show that the 5-kb (phase I) HapMap has sufficient density to enable linkage disequilibrium mapping in humans. Such studies will significantly enhance our ability to annotate the non-coding part of the genome and interpret functional variation. In addition, we demonstrate that the HapMap cell lines themselves may serve as a useful resource for quantitative measurements at the cellular level.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures
Similar articles
-
Perspectives on human genetic variation from the HapMap Project.PLoS Genet. 2005 Oct;1(4):e54. doi: 10.1371/journal.pgen.0010054. PLoS Genet. 2005. PMID: 16254603 Free PMC article.
-
Using haplotype blocks to map human complex trait loci.Trends Genet. 2003 Mar;19(3):135-40. doi: 10.1016/S0168-9525(03)00022-2. Trends Genet. 2003. PMID: 12615007 Review.
-
Fine-scale map of encyclopedia of DNA elements regions in the Korean population.Genetics. 2006 Sep;174(1):491-7. doi: 10.1534/genetics.105.052225. Epub 2006 May 15. Genetics. 2006. PMID: 16702437 Free PMC article.
-
Effects of single SNPs, haplotypes, and whole-genome LD maps on accuracy of association mapping.Genet Epidemiol. 2007 Apr;31(3):179-88. doi: 10.1002/gepi.20199. Genet Epidemiol. 2007. PMID: 17285621
-
Cis-acting regulatory variation in the human genome.Science. 2004 Oct 22;306(5696):647-50. doi: 10.1126/science.1101659. Science. 2004. PMID: 15499010 Review.
Cited by
-
Genome-wide survey of interindividual differences of RNA stability in human lymphoblastoid cell lines.Sci Rep. 2013;3:1318. doi: 10.1038/srep01318. Sci Rep. 2013. PMID: 23422947 Free PMC article.
-
Population differences in transcript-regulator expression quantitative trait loci.PLoS One. 2012;7(3):e34286. doi: 10.1371/journal.pone.0034286. Epub 2012 Mar 27. PLoS One. 2012. PMID: 22479588 Free PMC article.
-
An integrated network of microRNA and gene expression in ovarian cancer.BMC Bioinformatics. 2015;16 Suppl 5(Suppl 5):S5. doi: 10.1186/1471-2105-16-S5-S5. Epub 2015 Mar 18. BMC Bioinformatics. 2015. PMID: 25860109 Free PMC article.
-
Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci.BMC Genomics. 2015 Feb 27;16(1):138. doi: 10.1186/s12864-015-1292-z. BMC Genomics. 2015. PMID: 25766683 Free PMC article.
-
Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid.Genetics. 2006 Aug;173(4):2199-210. doi: 10.1534/genetics.106.060699. Epub 2006 May 15. Genetics. 2006. PMID: 16702414 Free PMC article.
References
-
- Storey JD, Akey JM, Kruglyak L. Multiple locus linkage analysis of genome-wide expression in yeast. PLoS Biol. 2005;3:e267. DOI: 10.1371/journal.pbio.0030267. - DOI - PMC - PubMed
-
- Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. - PubMed
-
- Yvert G, Brem RB, Whittle J, Akey JM, Foss E, et al. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet. 2003;35:57–64. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
