

Assembly and disassembly of nucleosome core particles containing histone variants by human nucleosome assembly protein I
- PMID: 16287874
- PMCID: PMC1291234
- DOI: 10.1128/MCB.25.23.10639-10651.2005
Assembly and disassembly of nucleosome core particles containing histone variants by human nucleosome assembly protein I
Abstract
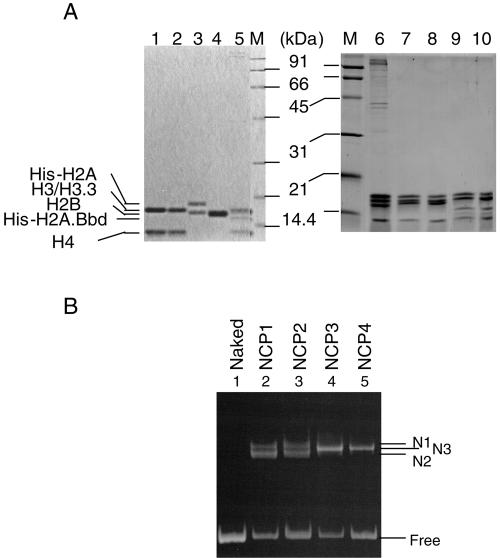
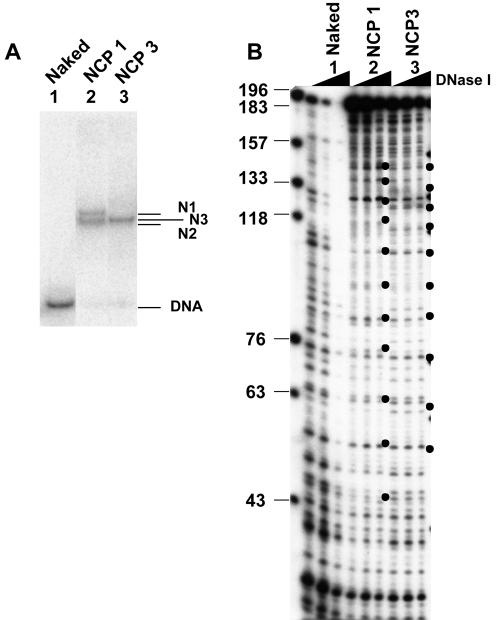
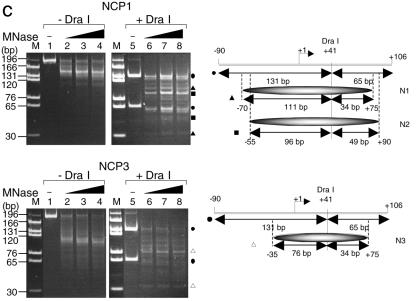
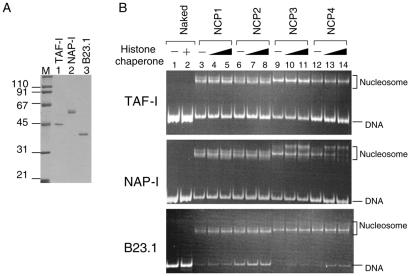
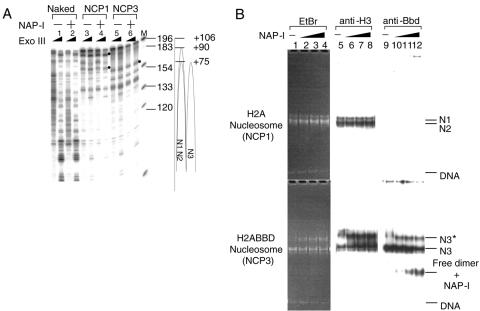
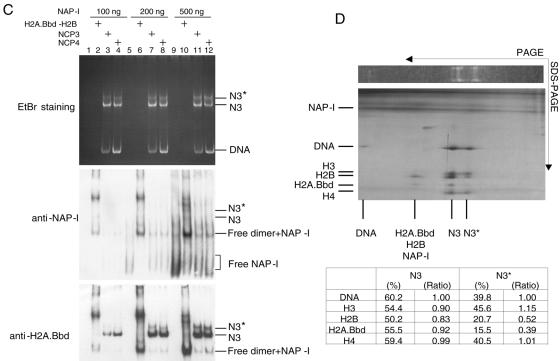
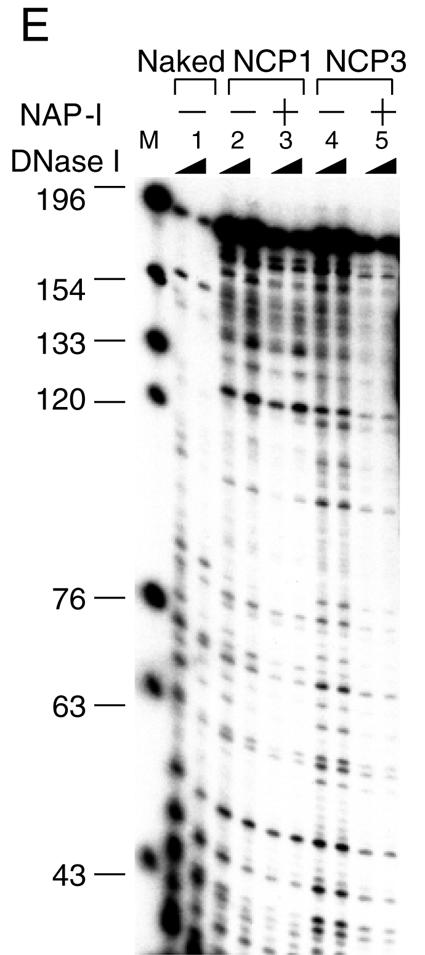
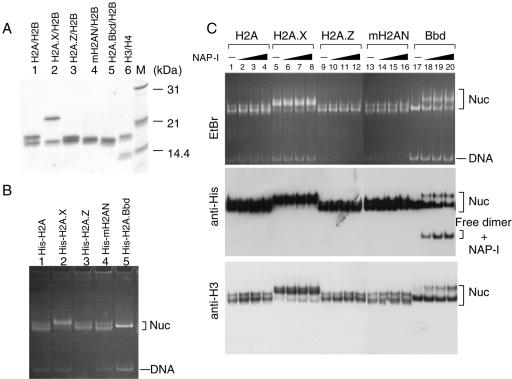
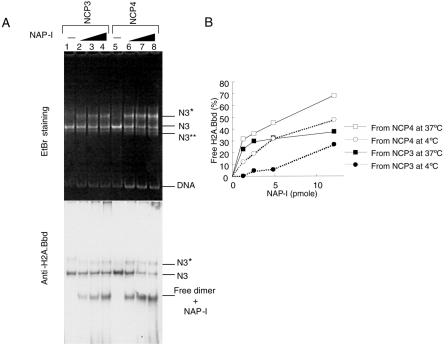
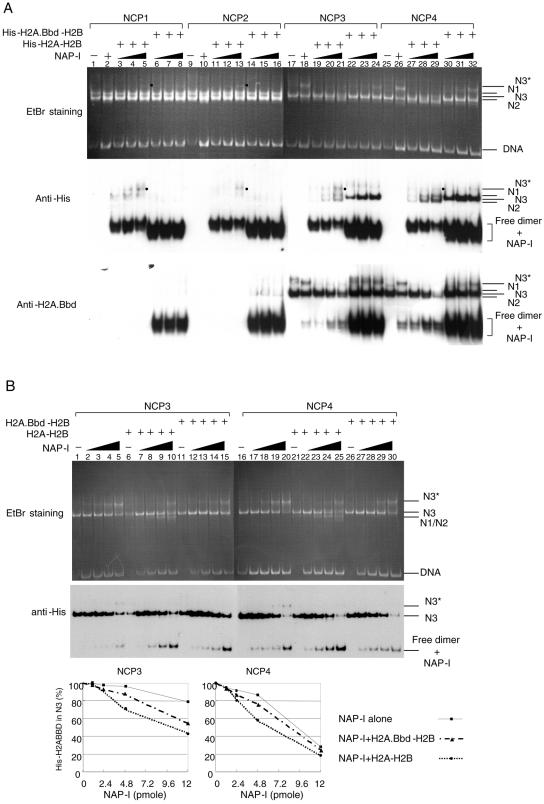
Histone variants play important roles in the maintenance and regulation of the chromatin structure. In order to characterize the biochemical properties of the chromatin structure containing histone variants, we investigated the dynamic status of nucleosome core particles (NCPs) that were assembled with recombinant histones. We found that in the presence of nucleosome assembly protein I (NAP-I), a histone chaperone, H2A-Barr body deficient (H2A.Bbd) confers the most flexible nucleosome structure among the mammalian histone H2A variants known thus far. NAP-I mediated the efficient assembly and disassembly of the H2A.Bbd-H2B dimers from NCPs. This reaction was accomplished more efficiently when the NCPs contained H3.3, a histone H3 variant known to be localized in the active chromatin, than when the NCPs contained the canonical H3. These observations indicate that the histone variants H2A.Bbd and H3.3 are involved in the formation and maintenance of the active chromatin structure. We also observed that acidic histone binding proteins, TAF-I/SET and B23.1, demonstrated dimer assembly and disassembly activity, but the efficiency of their activity was considerably lower than that of NAP-I. Thus, both the acidic nature of NAP-I and its other functional structure(s) may be essential to mediate the assembly and disassembly of the dimers in NCPs.
Figures
Similar articles
-
p300-mediated acetylation facilitates the transfer of histone H2A-H2B dimers from nucleosomes to a histone chaperone.Genes Dev. 2000 Aug 1;14(15):1899-907. Genes Dev. 2000. PMID: 10921904 Free PMC article.
-
H2A.Z and H3.3 histone variants affect nucleosome structure: biochemical and biophysical studies.Biochemistry. 2009 Nov 24;48(46):10852-7. doi: 10.1021/bi901129e. Biochemistry. 2009. PMID: 19856965
-
Functional characterization of human nucleosome assembly protein 1-like proteins as histone chaperones.Genes Cells. 2010 Jan;15(1):13-27. doi: 10.1111/j.1365-2443.2009.01361.x. Epub 2009 Dec 9. Genes Cells. 2010. PMID: 20002496
-
Quickly evolving histones, nucleosome stability and chromatin folding: all about histone H2A.Bbd.Gene. 2008 Apr 30;413(1-2):1-7. doi: 10.1016/j.gene.2008.02.003. Epub 2008 Feb 16. Gene. 2008. PMID: 18329190 Review.
-
Chromatin assembly: a basic recipe with various flavours.Curr Opin Genet Dev. 2006 Apr;16(2):104-11. doi: 10.1016/j.gde.2006.02.011. Epub 2006 Feb 28. Curr Opin Genet Dev. 2006. PMID: 16504499 Review.
Cited by
-
Structural basis of a nucleosome containing histone H2A.B/H2A.Bbd that transiently associates with reorganized chromatin.Sci Rep. 2013 Dec 16;3:3510. doi: 10.1038/srep03510. Sci Rep. 2013. PMID: 24336483 Free PMC article.
-
Roles of Histone H2A Variants in Cancer Development, Prognosis, and Treatment.Int J Mol Sci. 2024 Mar 9;25(6):3144. doi: 10.3390/ijms25063144. Int J Mol Sci. 2024. PMID: 38542118 Free PMC article. Review.
-
Histone variant H2A.B-H2B dimers are spontaneously exchanged with canonical H2A-H2B in the nucleosome.Commun Biol. 2021 Feb 12;4(1):191. doi: 10.1038/s42003-021-01707-z. Commun Biol. 2021. PMID: 33580188 Free PMC article.
-
Histone H2A variants in nucleosomes and chromatin: more or less stable?Nucleic Acids Res. 2012 Nov;40(21):10719-41. doi: 10.1093/nar/gks865. Epub 2012 Sep 21. Nucleic Acids Res. 2012. PMID: 23002134 Free PMC article. Review.
-
Nucleosome formation activity of human somatic nuclear autoantigenic sperm protein (sNASP).J Biol Chem. 2010 Apr 16;285(16):11913-21. doi: 10.1074/jbc.M109.083238. Epub 2010 Feb 18. J Biol Chem. 2010. PMID: 20167597 Free PMC article.
References
-
- Ahmad, K., and S. Henikoff. 2002. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9:1191-1200. - PubMed
-
- Akey, C. W., and K. Luger. 2003. Histone chaperones and nucleosome assembly. Curr. Opin. Struct. Biol. 13:6-14. - PubMed
-
- Belotserkovskaya, R., S. Oh, V. A. Bondarenko, G. Orphanides, V. M. Studitsky, and D. Reinberg. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301:1090-1093. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous