

Structural characterization of proteins using residue environments
- PMID: 16245324
- PMCID: PMC2483305
- DOI: 10.1002/prot.20661
Structural characterization of proteins using residue environments
Abstract
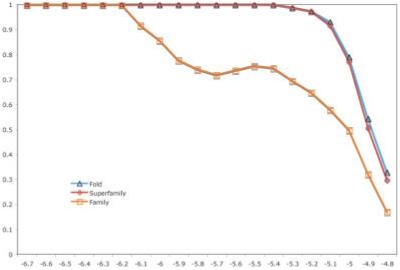
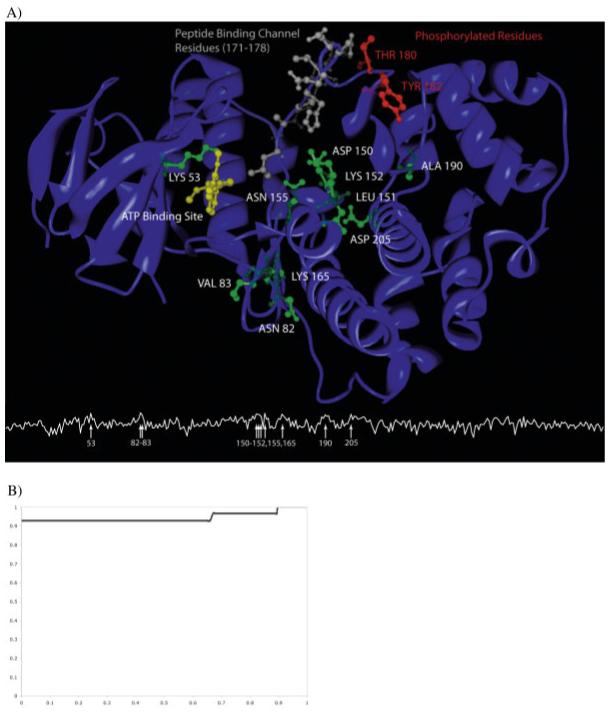
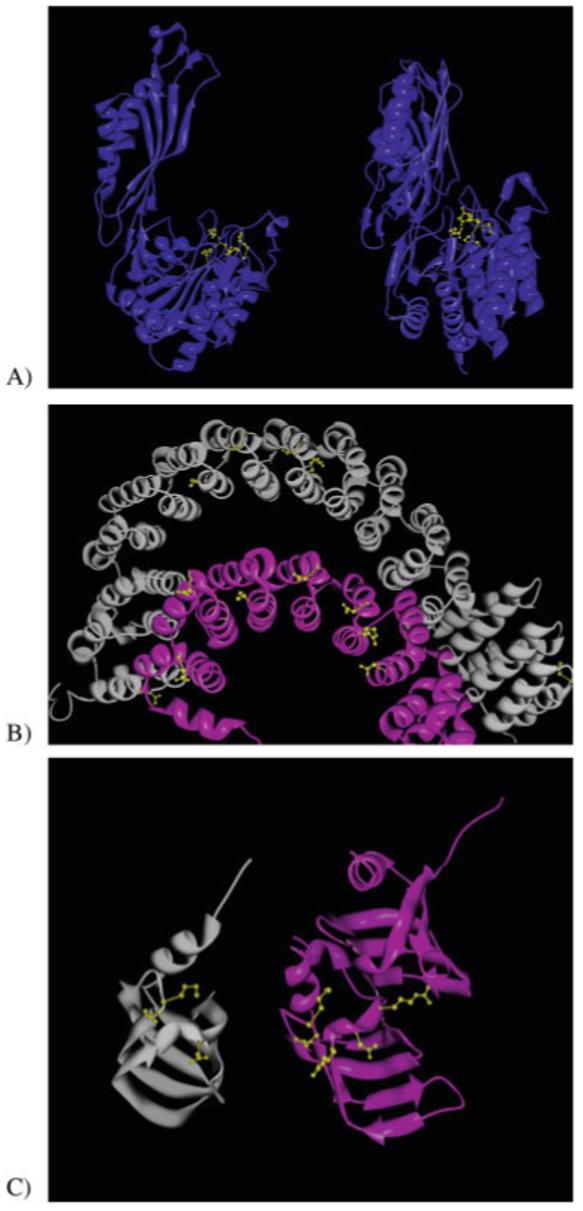
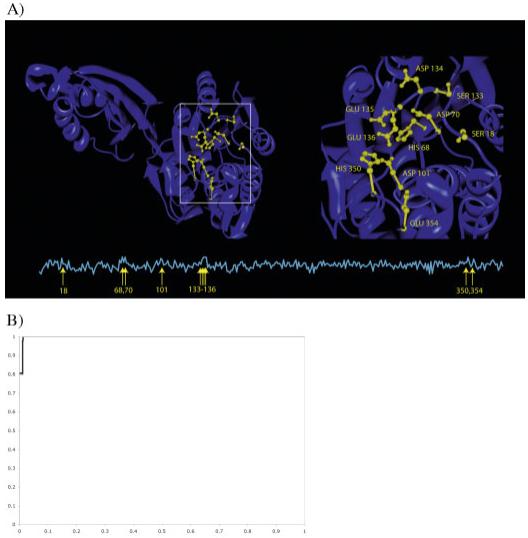
A primary challenge for structural genomics is the automated functional characterization of protein structures. We have developed a sequence-independent method called S-BLEST (Structure-Based Local Environment Search Tool) for the annotation of previously uncharacterized protein structures. S-BLEST encodes the local environment of an amino acid as a vector of structural property values. It has been applied to all amino acids in a nonredundant database of protein structures to generate a searchable structural resource. Given a query amino acid from an experimentally determined or modeled structure, S-BLEST quickly identifies similar amino acid environments using a K-nearest neighbor search. In addition, the method gives an estimation of the statistical significance of each result. We validated S-BLEST on X-ray crystal structures from the ASTRAL 40 nonredundant dataset. We then applied it to 86 crystallographically determined proteins in the protein data bank (PDB) with unknown function and with no significant sequence neighbors in the PDB. S-BLEST was able to associate 20 proteins with at least one local structural neighbor and identify the amino acid environments that are most similar between those neighbors.
Proteins 2005. 2005 Wiley-Liss, Inc.
Figures
Similar articles
-
Identification of similar regions of protein structures using integrated sequence and structure analysis tools.BMC Struct Biol. 2006 Mar 9;6:4. doi: 10.1186/1472-6807-6-4. BMC Struct Biol. 2006. PMID: 16526955 Free PMC article.
-
NdPASA: a novel pairwise protein sequence alignment algorithm that incorporates neighbor-dependent amino acid propensities.Proteins. 2005 Feb 15;58(3):628-37. doi: 10.1002/prot.20359. Proteins. 2005. PMID: 15616964
-
Intrinsic disorder in the Protein Data Bank.J Biomol Struct Dyn. 2007 Feb;24(4):325-42. doi: 10.1080/07391102.2007.10507123. J Biomol Struct Dyn. 2007. PMID: 17206849
-
The construction of an amino acid network for understanding protein structure and function.Amino Acids. 2014 Jun;46(6):1419-39. doi: 10.1007/s00726-014-1710-6. Epub 2014 Mar 13. Amino Acids. 2014. PMID: 24623120 Review.
-
Amino acid modifications for conformationally constraining naturally occurring and engineered peptide backbones: Insights from the Protein Data Bank.Biopolymers. 2018 Aug;109(10):e23230. doi: 10.1002/bip.23230. Epub 2018 Oct 25. Biopolymers. 2018. PMID: 30368772 Review.
Cited by
-
Protein structure prediction using residue- and fragment-environment potentials in CASP11.Proteins. 2016 Sep;84 Suppl 1(Suppl 1):105-17. doi: 10.1002/prot.24920. Epub 2015 Sep 22. Proteins. 2016. PMID: 26344195 Free PMC article.
-
The unfoldomics decade: an update on intrinsically disordered proteins.BMC Genomics. 2008 Sep 16;9 Suppl 2(Suppl 2):S1. doi: 10.1186/1471-2164-9-S2-S1. BMC Genomics. 2008. PMID: 18831774 Free PMC article.
-
Identification of similar regions of protein structures using integrated sequence and structure analysis tools.BMC Struct Biol. 2006 Mar 9;6:4. doi: 10.1186/1472-6807-6-4. BMC Struct Biol. 2006. PMID: 16526955 Free PMC article.
-
Graphlet kernels for prediction of functional residues in protein structures.J Comput Biol. 2010 Jan;17(1):55-72. doi: 10.1089/cmb.2009.0029. J Comput Biol. 2010. PMID: 20078397 Free PMC article.
-
Identification of recurring protein structure microenvironments and discovery of novel functional sites around CYS residues.BMC Struct Biol. 2010 Feb 2;10:4. doi: 10.1186/1472-6807-10-4. BMC Struct Biol. 2010. PMID: 20122268 Free PMC article.
References
-
- Thornton JM, Todd AE, Milburn D, Borkakoti N, Orengo CA. From structure to function: approaches and limitations. Nat Struct Biol. 2000;7(Suppl):991–994. - PubMed
-
- Lichtarge O, Bourne H, Cohen F. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257(2):342–358. - PubMed
-
- Fetrow J, Skolnick J. Method for prediction of protein function from sequence using the sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J Mol Bioly. 1998;281:949–968. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
