

beta-Adrenoreceptors reactivate Kaposi's sarcoma-associated herpesvirus lytic replication via PKA-dependent control of viral RTA
- PMID: 16227274
- PMCID: PMC1262578
- DOI: 10.1128/JVI.79.21.13538-13547.2005
beta-Adrenoreceptors reactivate Kaposi's sarcoma-associated herpesvirus lytic replication via PKA-dependent control of viral RTA
Abstract
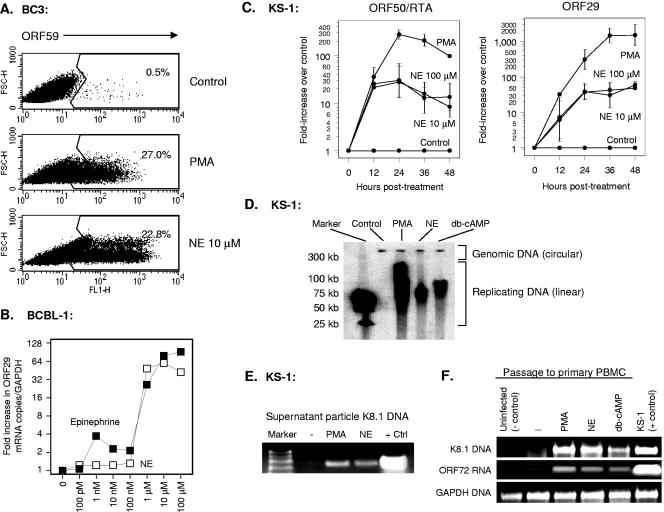
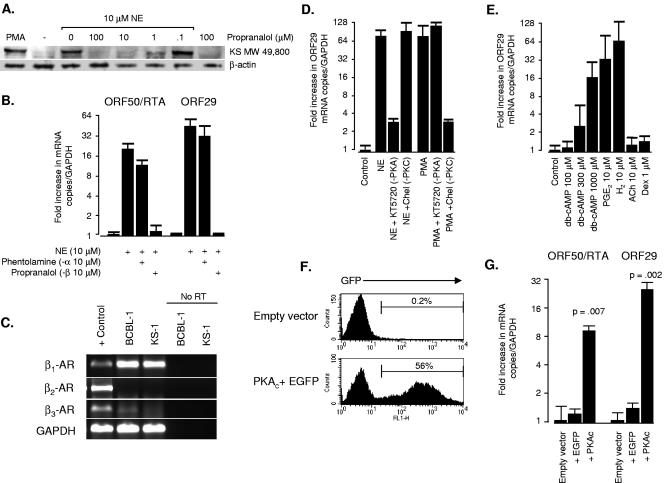
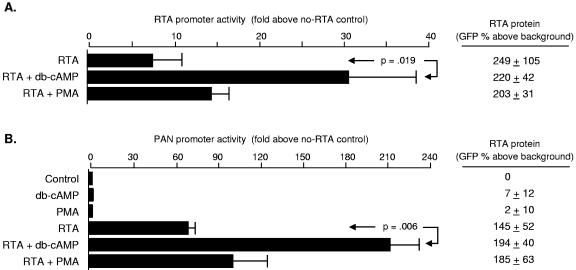
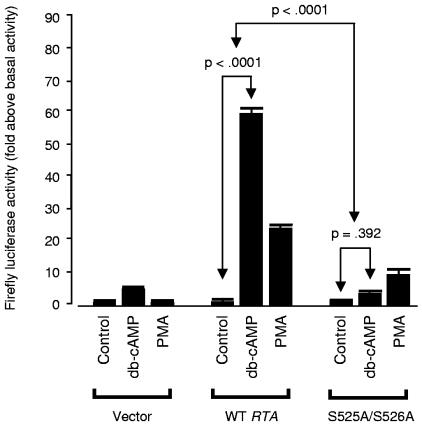
Reactivation of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic replication is mediated by the viral RTA transcription factor, but little is known about the physiological processes controlling its expression or activity. Links between autonomic nervous system activity and AIDS-associated Kaposi's sarcoma led us to examine the potential influence of catecholamine neurotransmitters. Physiological concentrations of epinephrine and norepinephrine efficiently reactivated lytic replication of KSHV in latently infected primary effusion lymphoma cells via beta-adrenergic activation of the cellular cyclic AMP/protein kinase A (PKA) signaling pathway. Effects were blocked by PKA antagonists and mimicked by pharmacological and physiological PKA activators (prostaglandin E2 and histamine) or overexpression of the PKA catalytic subunit. PKA up-regulated RTA gene expression, enhanced activity of the RTA promoter, and posttranslationally enhanced RTA's trans-activating capacity for its own promoter and heterologous lytic promoters (e.g., the viral PAN gene). Mutation of predicted phosphorylation targets at RTA serines 525 and 526 inhibited PKA-mediated enhancement of RTA trans-activating capacity. Given the high catecholamine levels at sites of KSHV latency such as the vasculature and lymphoid organs, these data suggest that beta-adrenergic control of RTA might constitute a significant physiological regulator of KSHV lytic replication. These findings also suggest novel therapeutic strategies for controlling the activity of this oncogenic gammaherpesvirus in vivo.
Figures

Similar articles
-
KSHV reactivation and novel implications of protein isomerization on lytic switch control.Viruses. 2015 Jan 12;7(1):72-109. doi: 10.3390/v7010072. Viruses. 2015. PMID: 25588053 Free PMC article. Review.
-
Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency.J Virol. 2004 Jun;78(12):6585-94. doi: 10.1128/JVI.78.12.6585-6594.2004. J Virol. 2004. PMID: 15163750 Free PMC article.
-
Activation of Kaposi's sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: identification of sirtuin 1 as a regulator of the KSHV life cycle.J Virol. 2014 Jun;88(11):6355-67. doi: 10.1128/JVI.00219-14. Epub 2014 Mar 26. J Virol. 2014. PMID: 24672028 Free PMC article.
-
Identification of Novel Kaposi's Sarcoma-Associated Herpesvirus Orf50 Transcripts: Discovery of New RTA Isoforms with Variable Transactivation Potential.J Virol. 2016 Dec 16;91(1):e01434-16. doi: 10.1128/JVI.01434-16. Print 2017 Jan 1. J Virol. 2016. PMID: 27795414 Free PMC article.
-
The Rta/Orf50 transactivator proteins of the gamma-herpesviridae.Curr Top Microbiol Immunol. 2007;312:71-100. doi: 10.1007/978-3-540-34344-8_3. Curr Top Microbiol Immunol. 2007. PMID: 17089794 Review.
Cited by
-
Failed Treatment of Classic Kaposi's Sarcoma with Topical Timolol: Case Report and Review of the Literature.Cureus. 2019 Dec 2;11(12):e6272. doi: 10.7759/cureus.6272. Cureus. 2019. PMID: 31903308 Free PMC article.
-
KSHV reactivation and novel implications of protein isomerization on lytic switch control.Viruses. 2015 Jan 12;7(1):72-109. doi: 10.3390/v7010072. Viruses. 2015. PMID: 25588053 Free PMC article. Review.
-
Viral latency and its regulation: lessons from the gamma-herpesviruses.Cell Host Microbe. 2010 Jul 22;8(1):100-15. doi: 10.1016/j.chom.2010.06.014. Cell Host Microbe. 2010. PMID: 20638646 Free PMC article. Review.
-
Control of Kaposi's sarcoma-associated herpesvirus reactivation induced by multiple signals.PLoS One. 2011;6(6):e20998. doi: 10.1371/journal.pone.0020998. Epub 2011 Jun 28. PLoS One. 2011. PMID: 21904595 Free PMC article.
-
Nervous system regulation of the cancer genome.Brain Behav Immun. 2013 Mar;30 Suppl(Suppl):S10-8. doi: 10.1016/j.bbi.2012.11.008. Epub 2012 Dec 1. Brain Behav Immun. 2013. PMID: 23207104 Free PMC article. Review.
References
-
- Bloom, D. C., J. G. Stevens, J. M. Hill, and R. K. Tran. 1997. Mutagenesis of a cAMP response element within the latency-associated transcript promoter of HSV-1 reduces adrenergic reactivation. Virology 236:202-207. - PubMed
-
- Cacioppo, J. T., J. K. Kiecolt-Glaser, W. B. Malarkey, B. F. Laskowski, L. A. Rozlog, K. M. Poehlmann, M. H. Burleson, and R. Glaser. 2002. Autonomic and glucocorticoid associations with the steady-state expression of latent Epstein-Barr virus. Horm. Behav. 42:32-41. - PubMed
-
- Carey, M., and S. T. Smale. 2000. Transcriptional regulation in eukaryotes: concepts, strategies, and techniques. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
