

The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways
- PMID: 16177794
- PMCID: PMC1636584
- DOI: 10.1038/nature03966
The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways
Abstract
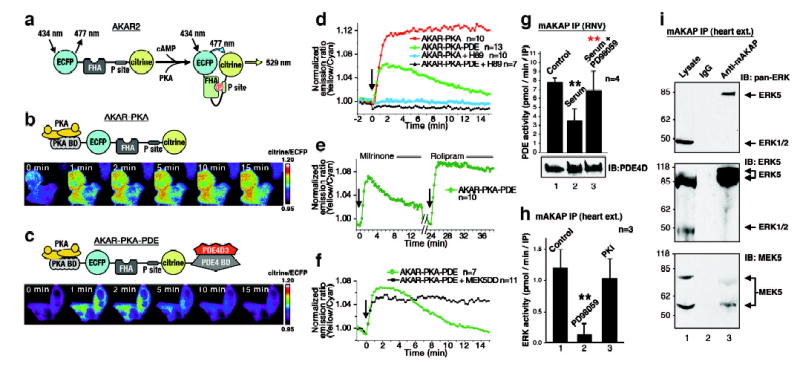
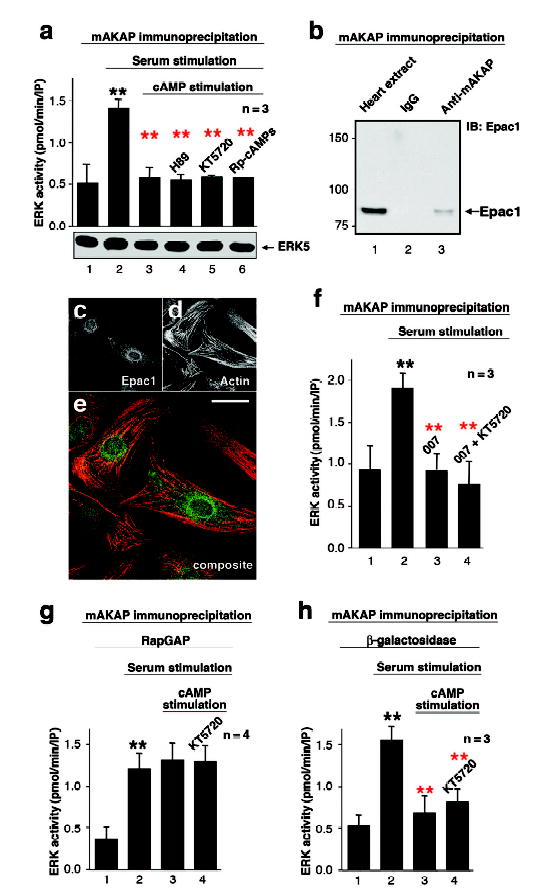
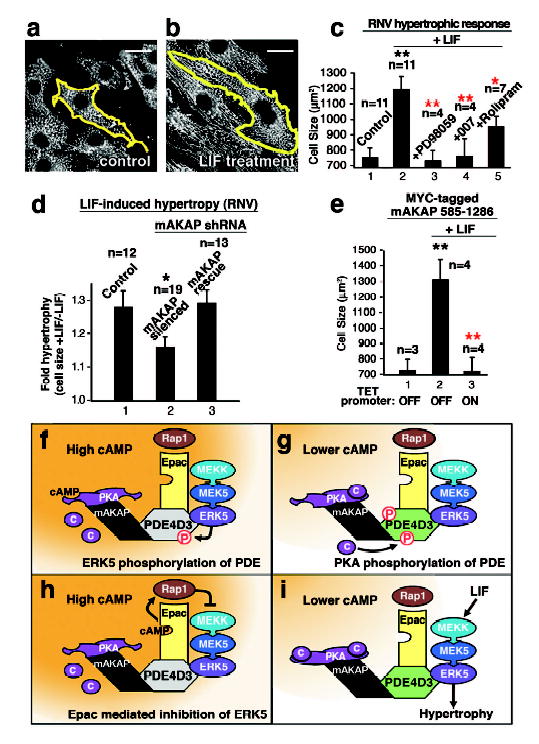
Cyclic adenosine 3', 5'-monophosphate (cAMP) is a ubiquitous mediator of intracellular signalling events. It acts principally through stimulation of cAMP-dependent protein kinases (PKAs) but also activates certain ion channels and guanine nucleotide exchange factors (Epacs). Metabolism of cAMP is catalysed by phosphodiesterases (PDEs). Here we identify a cAMP-responsive signalling complex maintained by the muscle-specific A-kinase anchoring protein (mAKAP) that includes PKA, PDE4D3 and Epac1. These intermolecular interactions facilitate the dissemination of distinct cAMP signals through each effector protein. Anchored PKA stimulates PDE4D3 to reduce local cAMP concentrations, whereas an mAKAP-associated ERK5 kinase module suppresses PDE4D3. PDE4D3 also functions as an adaptor protein that recruits Epac1, an exchange factor for the small GTPase Rap1, to enable cAMP-dependent attenuation of ERK5. Pharmacological and molecular manipulations of the mAKAP complex show that anchored ERK5 can induce cardiomyocyte hypertrophy. Thus, two coupled cAMP-dependent feedback loops are coordinated within the context of the mAKAP complex, suggesting that local control of cAMP signalling by AKAP proteins is more intricate than previously appreciated.
Conflict of interest statement
Competing Interests statement The authors declare that there are no competing financial interests.
Figures
Similar articles
-
mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module.EMBO J. 2001 Apr 17;20(8):1921-30. doi: 10.1093/emboj/20.8.1921. EMBO J. 2001. PMID: 11296225 Free PMC article.
-
The mAKAP signaling complex: integration of cAMP, calcium, and MAP kinase signaling pathways.Eur J Cell Biol. 2006 Jul;85(7):593-602. doi: 10.1016/j.ejcb.2006.01.007. Epub 2006 Feb 7. Eur J Cell Biol. 2006. PMID: 16460834 Review.
-
PKA-phosphorylation of PDE4D3 facilitates recruitment of the mAKAP signalling complex.Biochem J. 2004 Aug 1;381(Pt 3):587-92. doi: 10.1042/BJ20040846. Biochem J. 2004. PMID: 15182229 Free PMC article.
-
AKAP signaling complexes: getting to the heart of the matter.Trends Mol Med. 2006 Jul;12(7):317-23. doi: 10.1016/j.molmed.2006.05.008. Epub 2006 Jun 30. Trends Mol Med. 2006. PMID: 16809066 Review.
-
cAMP-stimulated protein phosphatase 2A activity associated with muscle A kinase-anchoring protein (mAKAP) signaling complexes inhibits the phosphorylation and activity of the cAMP-specific phosphodiesterase PDE4D3.J Biol Chem. 2010 Apr 9;285(15):11078-86. doi: 10.1074/jbc.M109.034868. Epub 2010 Jan 27. J Biol Chem. 2010. PMID: 20106966 Free PMC article.
Cited by
-
The role of Epac in the heart.Cell Mol Life Sci. 2017 Feb;74(4):591-606. doi: 10.1007/s00018-016-2336-5. Epub 2016 Aug 22. Cell Mol Life Sci. 2017. PMID: 27549789 Free PMC article. Review.
-
PKA regulatory subunits mediate synergy among conserved G-protein-coupled receptor cascades.Nat Commun. 2011 Dec 20;2:598. doi: 10.1038/ncomms1605. Nat Commun. 2011. PMID: 22186894 Free PMC article.
-
Mechanisms of action of glucagon-like peptide 1 in the pancreas.Pharmacol Ther. 2007 Mar;113(3):546-93. doi: 10.1016/j.pharmthera.2006.11.007. Epub 2006 Dec 28. Pharmacol Ther. 2007. PMID: 17306374 Free PMC article. Review.
-
Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart.Circ Res. 2007 Mar 2;100(4):489-501. doi: 10.1161/01.RES.0000258451.44949.d7. Circ Res. 2007. PMID: 17332439 Free PMC article. Review.
-
The mAKAPbeta scaffold regulates cardiac myocyte hypertrophy via recruitment of activated calcineurin.J Mol Cell Cardiol. 2010 Feb;48(2):387-94. doi: 10.1016/j.yjmcc.2009.10.023. Epub 2009 Oct 31. J Mol Cell Cardiol. 2010. PMID: 19883655 Free PMC article.
References
-
- Walsh DA, Perkins JP, Krebs EG. An adenosine 3′,5′-monophosphate-dependent protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763–3765. - PubMed
-
- Su Y, et al. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding proteins. Science. 1995;269:807–813. - PubMed
-
- Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–8. - PubMed
-
- Beavo JA, Brunton LL. Cyclic nucleotide research -- still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
