

Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease
- PMID: 16172208
- PMCID: PMC2171517
- DOI: 10.1083/jcb.200507087
Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease
Abstract
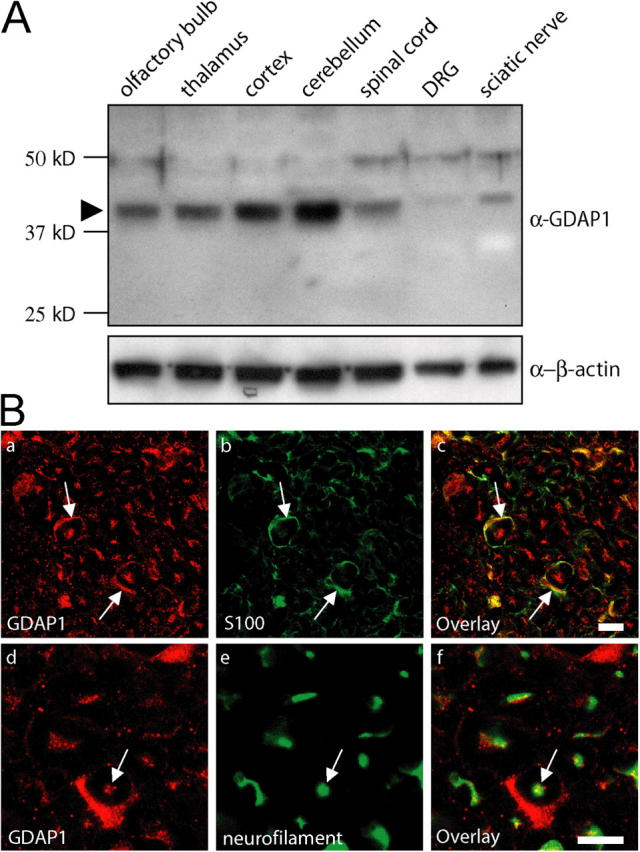
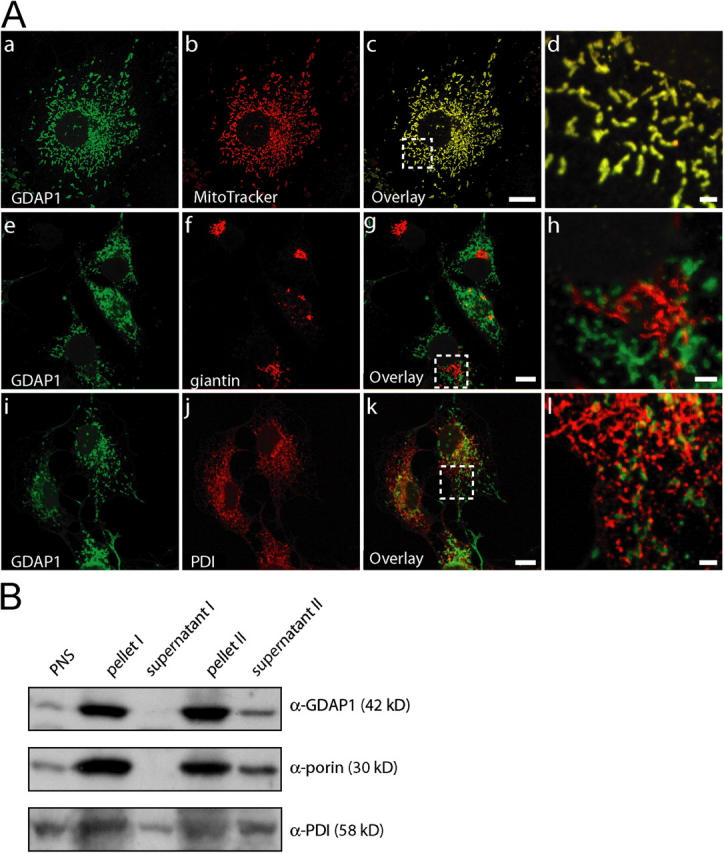
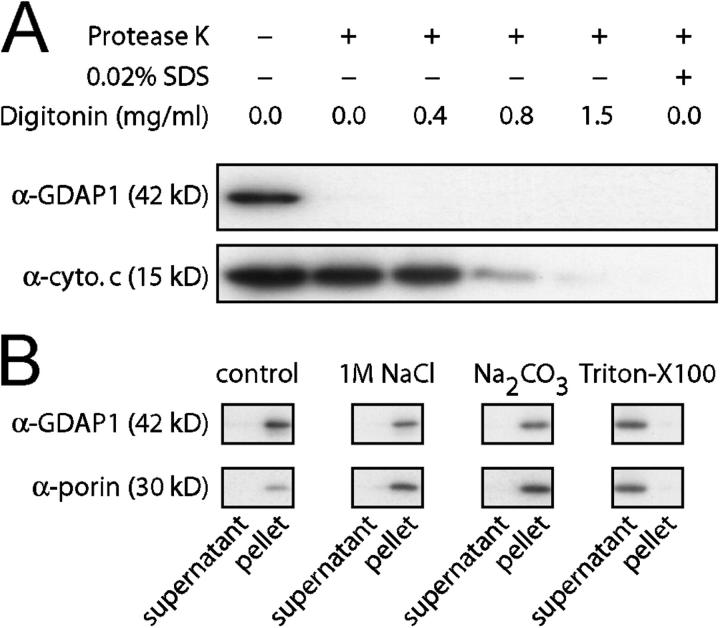
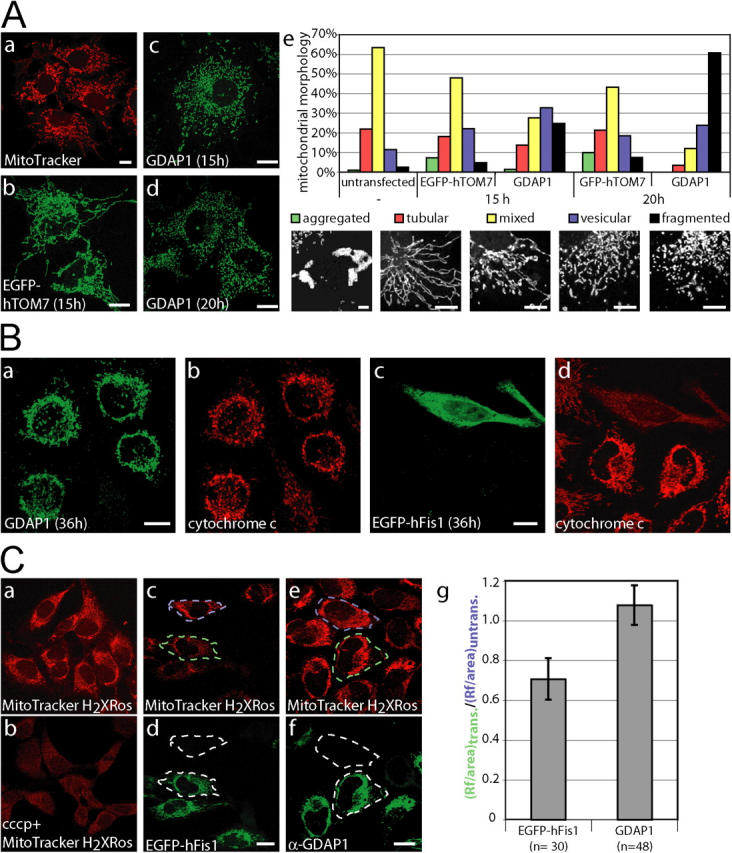
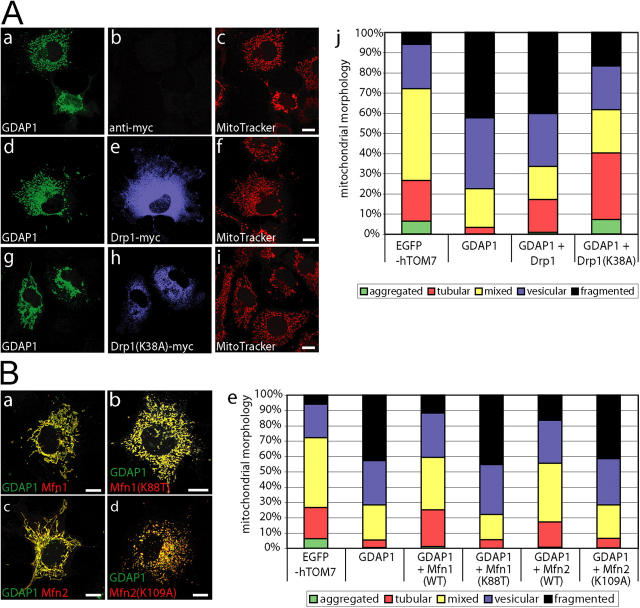
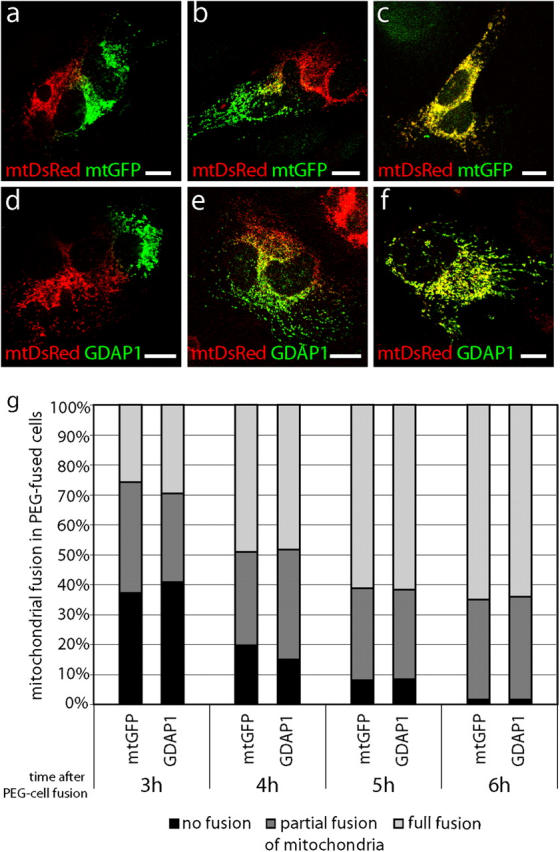
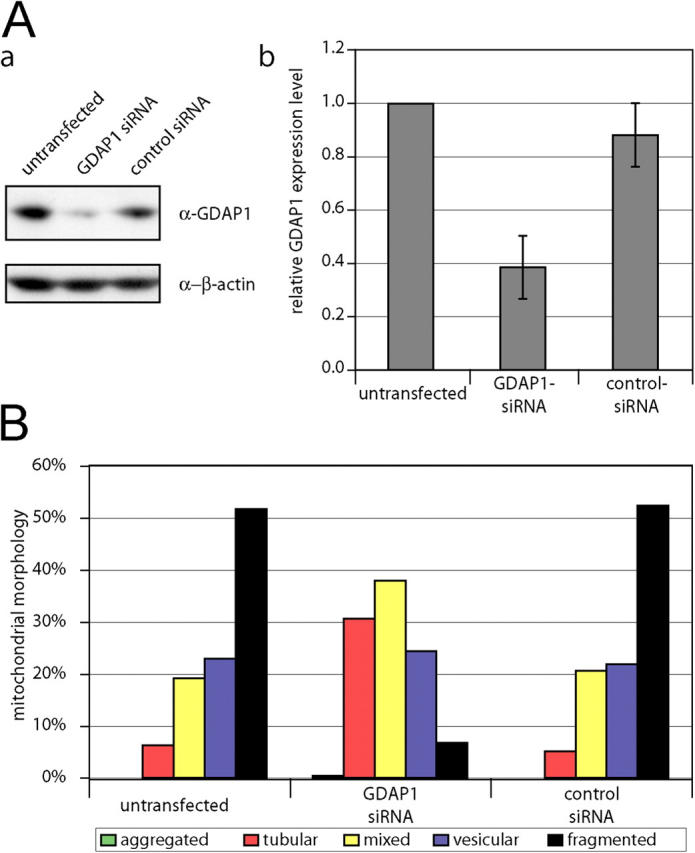
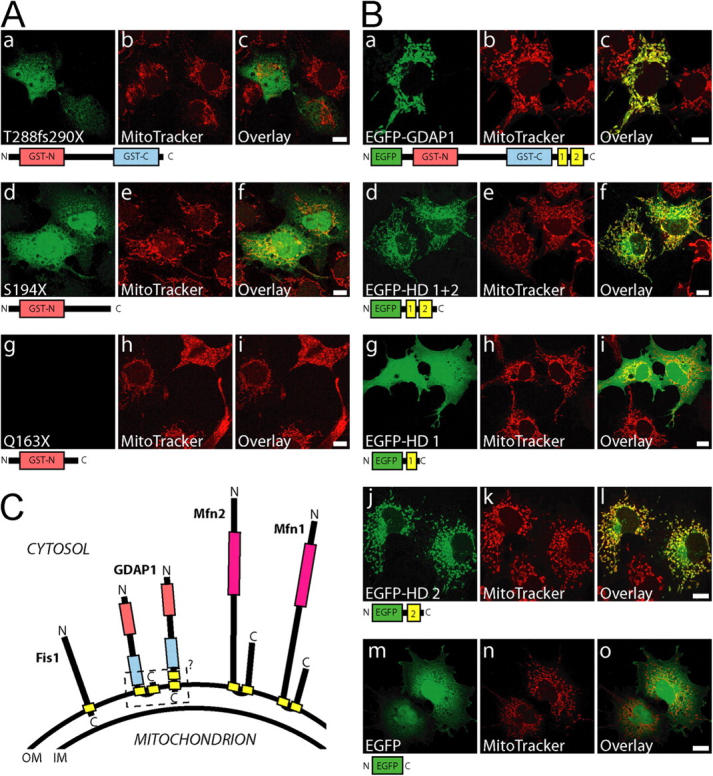
Mutations in GDAP1 lead to severe forms of the peripheral motor and sensory neuropathy, Charcot-Marie-Tooth disease (CMT), which is characterized by heterogeneous phenotypes, including pronounced axonal damage and demyelination. We show that neurons and Schwann cells express ganglioside-induced differentiation associated protein 1 (GDAP1), which suggest that both cell types may contribute to the mixed features of the disease. GDAP1 is located in the mitochondrial outer membrane and regulates the mitochondrial network. Overexpression of GDAP1 induces fragmentation of mitochondria without inducing apoptosis, affecting overall mitochondrial activity, or interfering with mitochondrial fusion. The mitochondrial fusion proteins, mitofusin 1 and 2 and Drp1(K38A), can counterbalance the GDAP1-dependent fission. GDAP1-specific knockdown by RNA interference results in a tubular mitochondrial morphology. GDAP1 truncations that are found in patients who have CMT are not targeted to mitochondria and have lost mitochondrial fragmentation activity. The latter activity also is reduced strongly for disease-associated GDAP1 point mutations. Our data indicate that an exquisitely tight control of mitochondrial dynamics, regulated by GDAP1, is crucial for the proper function of myelinated peripheral nerves.
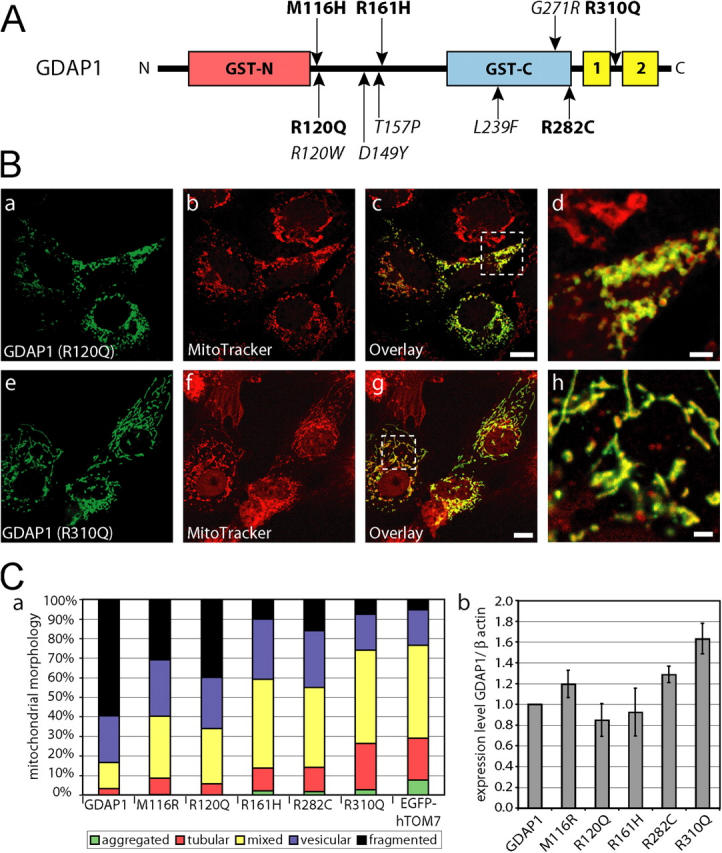
Figures
Similar articles
-
Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission.EMBO Rep. 2013 Jun;14(6):545-52. doi: 10.1038/embor.2013.56. Epub 2013 Apr 30. EMBO Rep. 2013. PMID: 23628762 Free PMC article.
-
The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease.Brain. 2014 Mar;137(Pt 3):668-82. doi: 10.1093/brain/awt371. Epub 2014 Jan 29. Brain. 2014. PMID: 24480485 Free PMC article.
-
Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot-Marie-Tooth type 4A disease.J Cell Mol Med. 2008 Apr;12(2):679-89. doi: 10.1111/j.1582-4934.2007.00158.x. Epub 2007 Nov 16. J Cell Mol Med. 2008. PMID: 18021315 Free PMC article.
-
Calcium Deregulation and Mitochondrial Bioenergetics in GDAP1-Related CMT Disease.Int J Mol Sci. 2019 Jan 18;20(2):403. doi: 10.3390/ijms20020403. Int J Mol Sci. 2019. PMID: 30669311 Free PMC article. Review.
-
Mitochondrial dynamics and inherited peripheral nerve diseases.Neurosci Lett. 2015 Jun 2;596:66-77. doi: 10.1016/j.neulet.2015.04.001. Epub 2015 Apr 3. Neurosci Lett. 2015. PMID: 25847151 Review.
Cited by
-
Lack of GDAP1 induces neuronal calcium and mitochondrial defects in a knockout mouse model of charcot-marie-tooth neuropathy.PLoS Genet. 2015 Apr 10;11(4):e1005115. doi: 10.1371/journal.pgen.1005115. eCollection 2015 Apr. PLoS Genet. 2015. PMID: 25860513 Free PMC article.
-
STAT6 in mitochondrial outer membrane impairs mitochondrial fusion by inhibiting MFN2 dimerization.iScience. 2022 Aug 13;25(9):104923. doi: 10.1016/j.isci.2022.104923. eCollection 2022 Sep 16. iScience. 2022. PMID: 36065189 Free PMC article.
-
Peroxisome-mitochondria interplay and disease.J Inherit Metab Dis. 2015 Jul;38(4):681-702. doi: 10.1007/s10545-015-9819-7. Epub 2015 Feb 17. J Inherit Metab Dis. 2015. PMID: 25687155 Review.
-
Regulation of mitochondrial dynamics: convergences and divergences between yeast and vertebrates.Cell Mol Life Sci. 2013 Mar;70(6):951-76. doi: 10.1007/s00018-012-1066-6. Epub 2012 Jul 18. Cell Mol Life Sci. 2013. PMID: 22806564 Free PMC article. Review.
-
New insights into the role of mitochondria in aging: mitochondrial dynamics and more.J Cell Sci. 2010 Aug 1;123(Pt 15):2533-42. doi: 10.1242/jcs.070490. J Cell Sci. 2010. PMID: 20940129 Free PMC article. Review.
References
-
- Ammar, N., E. Nelis, L. Merlini, N. Barisic, R. Amouri, C. Ceuterick, J.J. Martin, V. Timmerman, F. Hentati, and P. De Jonghe. 2003. Identification of novel GDAP1 mutations causing autosomal recessive Charcot-Marie-Tooth disease. Neuromuscul. Disord. 13:720–728. - PubMed
-
- Azzedine, H., M. Ruberg, D. Ente, C. Gilardeau, S. Perie, B. Wechsler, A. Brice, E. LeGuern, and O. Dubourg. 2003. Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul. Disord. 13:341–346. - PubMed
-
- Bach, D., S. Pich, F.X. Soriano, N. Vega, B. Baumgartner, J. Oriola, J.R. Daugaard, J. Lloberas, M. Camps, J.R. Zierath, et al. 2003. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 278:17190–17197. - PubMed
-
- Baxter, R.V., K. Ben Othmane, J.M. Rochelle, J.E. Stajich, C. Hulette, S. Dew-Knight, F. Hentati, M. Ben Hamida, S. Bel, J.E. Stenger, et al. 2002. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 30:21–22. - PubMed
-
- Berger, P., E.E. Sirkowski, S.S. Scherer, and U. Suter. 2004. Expression analysis of the N-Myc downstream-regulated gene 1 indicates that myelinating Schwann cells are the primary disease target in hereditary motor and sensory neuropathy-Lom. Neurobiol. Dis. 17:290–299. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
