

Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis
- PMID: 16166619
- PMCID: PMC2662988
- DOI: 10.1164/rccm.200504-644OC
Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis
Abstract
Rationale: Many of the interstitial lung diseases represent a diagnostic and therapeutic challenge because their clinical and even histologic features are often nonspecific. Likewise, the transcriptional signatures of most of them are unknown.
Objective: To compare the gene expression patterns from patients with idiopathic pulmonary fibrosis (IPF) hypersensitivity pneumonitis (HP), and nonspecific interstitial pneumonia (NSIP) using custom oligonucleotide microarrays.
Methods: We profiled lung biopsies from 15 patients with IPF, 12 with HP, and eight with NSIP. Labeled complementary ribonucleic acid was hybridized to a custom Affymetrix oligonucleotide DNA microarray using standard Affymetrix protocols. The custom array, Hu03, contained 59,619 probe sets representing an estimated 46,000 gene clusters.
Results: We identified statistically significant gene expression signatures that characterize HP and IPF. The HP gene expression signature was enriched for genes that are functionally associated with inflammation, T-cell activation, and immune responses, whereas the IPF signature was characterized by the expression of tissue remodeling, epithelial, and myofibroblast genes. We then compared these gene expression signatures to classify NSIP, a histologic pattern that is often difficult to differentiate consistently from HP and IPF. Two cases exhibited an IPF-like gene expression, another one could be more properly classified as HP, whereas others did not resemble HP or IPF, suggesting that they may represent idiopathic NSIP.
Conclusions: Our results underscore the value of gene expression signatures to classify the interstitial lung diseases and to understand pathogenic mechanisms, and suggest new ways to improve the diagnosis and treatment of patients with these diseases.
Figures
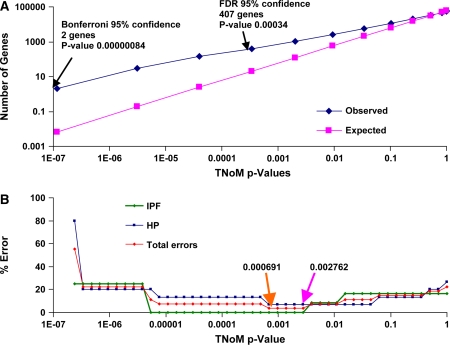
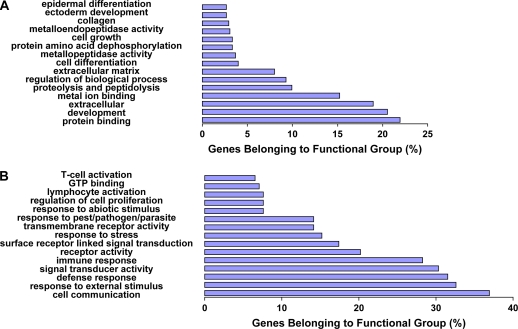
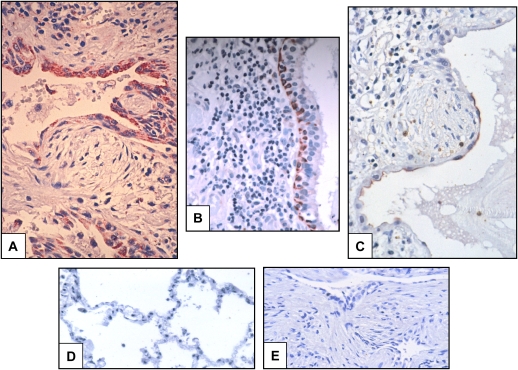


Comment in
-
Classification of interstitial pneumonias: what do gene expression profiles tell us?Am J Respir Crit Care Med. 2006 Jan 15;173(2):141-2. doi: 10.1164/rccm.2510004. Am J Respir Crit Care Med. 2006. PMID: 16391303 No abstract available.
Similar articles
-
Comprehensive gene expression profiling identifies distinct and overlapping transcriptional profiles in non-specific interstitial pneumonia and idiopathic pulmonary fibrosis.Respir Res. 2018 Aug 15;19(1):153. doi: 10.1186/s12931-018-0857-1. Respir Res. 2018. PMID: 30111332 Free PMC article.
-
Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT.Radiology. 2008 Jan;246(1):288-97. doi: 10.1148/radiol.2453061881. Radiology. 2008. PMID: 18096541
-
Role of surgical lung biopsy in separating chronic hypersensitivity pneumonia from usual interstitial pneumonia/idiopathic pulmonary fibrosis: analysis of 31 biopsies from 15 patients.Chest. 2008 Jul;134(1):126-32. doi: 10.1378/chest.08-0033. Epub 2008 Mar 13. Chest. 2008. PMID: 18339775
-
Pathology of hypersensitivity pneumonitis.Curr Opin Pulm Med. 2008 Sep;14(5):440-54. doi: 10.1097/MCP.0b013e3283043dfa. Curr Opin Pulm Med. 2008. PMID: 18664975 Review.
-
[Fibrotic hypersensitivity pneumonia: focus on pathology-relevant aspects of the new ATS/JRS/ALAT clinical guideline for the diagnosis of hypersensitivity pneumonia in adults].Pathologe. 2021 Feb;42(1):48-54. doi: 10.1007/s00292-020-00885-7. Epub 2020 Dec 23. Pathologe. 2021. PMID: 33355704 Free PMC article. Review. German.
Cited by
-
Lung cell transplantation for pulmonary fibrosis.Sci Adv. 2024 Aug 23;10(34):eadk2524. doi: 10.1126/sciadv.adk2524. Epub 2024 Aug 23. Sci Adv. 2024. PMID: 39178253 Free PMC article.
-
Rationale and design of the prognostic transcriptomic signature in fibrotic hypersensitivity pneumonitis (PREDICT) study.ERJ Open Res. 2024 Jan 8;10(1):00625-2023. doi: 10.1183/23120541.00625-2023. eCollection 2024 Jan. ERJ Open Res. 2024. PMID: 38264150 Free PMC article.
-
MMP-1 promoter polymorphism is associated with risk of radiation-induced lung injury in lung cancer patients treated with radiotherapy.Oncotarget. 2016 Oct 25;7(43):70175-70184. doi: 10.18632/oncotarget.12164. Oncotarget. 2016. PMID: 27659527 Free PMC article.
-
Altered DNA methylation profile in idiopathic pulmonary fibrosis.Am J Respir Crit Care Med. 2012 Sep 15;186(6):525-35. doi: 10.1164/rccm.201201-0077OC. Epub 2012 Jun 14. Am J Respir Crit Care Med. 2012. PMID: 22700861 Free PMC article.
-
Disease-specific gene expression profiling in multiple models of lung disease.Am J Respir Crit Care Med. 2008 Feb 15;177(4):376-87. doi: 10.1164/rccm.200702-333OC. Epub 2007 Nov 20. Am J Respir Crit Care Med. 2008. PMID: 18029791 Free PMC article.
References
-
- Schwarz ML, King TE, Raghu G. Approach to the evaluation and diagnosis of interstitial lung disease. In: King TE, Schwarz ML, editors. Interstitial lung disease. Ontario, BC, Canada: B.C. Decker; 2003. pp. 1–30.
-
- American Thoracic Society/European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002;165:277–304. - PubMed
-
- Katzenstein ALA, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998;157:1301–1315. - PubMed
-
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000;162:2213–2217. - PubMed
-
- Patel AM, Ryu JH, Reed CE. Hypersensitivity pneumonitis: current concepts and future questions. J Allergy Clin Immunol 2001;108:661–670. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
