

Multiple locus linkage analysis of genomewide expression in yeast
- PMID: 16035920
- PMCID: PMC1180512
- DOI: 10.1371/journal.pbio.0030267
Multiple locus linkage analysis of genomewide expression in yeast
Abstract
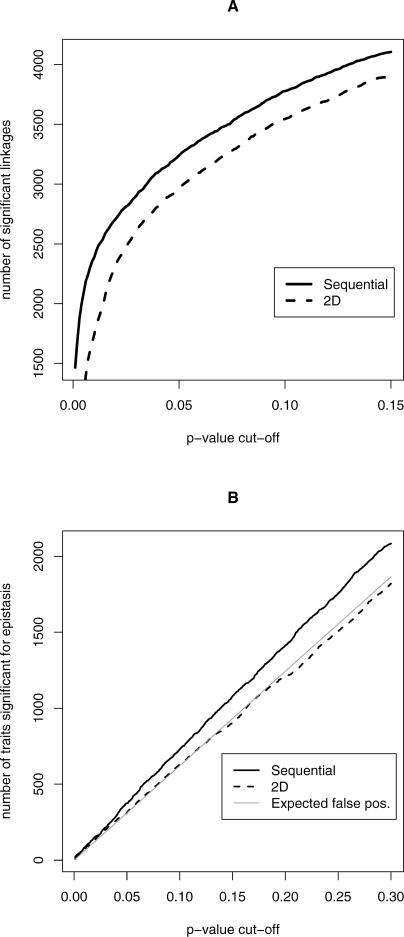
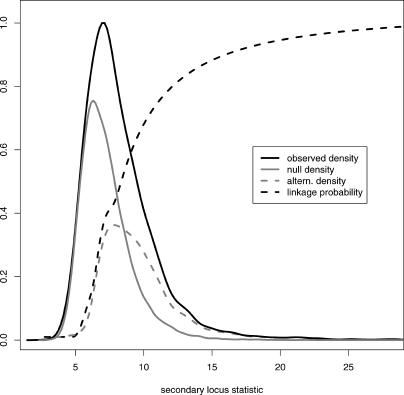
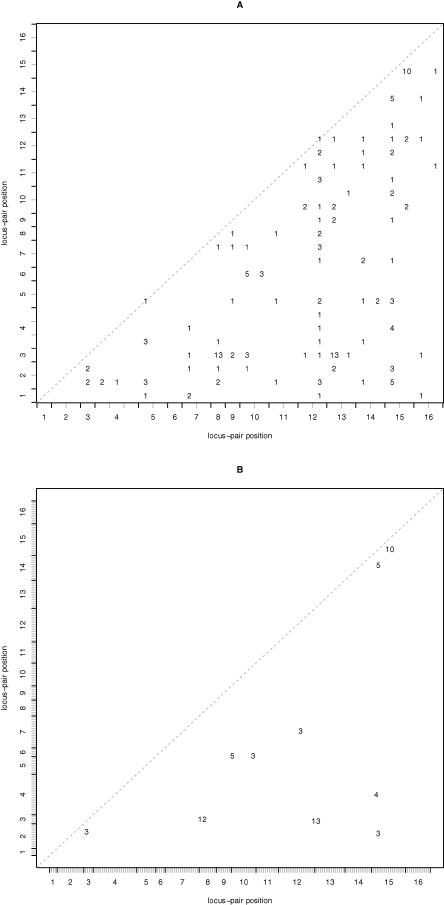
With the ability to measure thousands of related phenotypes from a single biological sample, it is now feasible to genetically dissect systems-level biological phenomena. The genetics of transcriptional regulation and protein abundance are likely to be complex, meaning that genetic variation at multiple loci will influence these phenotypes. Several recent studies have investigated the role of genetic variation in transcription by applying traditional linkage analysis methods to genomewide expression data, where each gene expression level was treated as a quantitative trait and analyzed separately from one another. Here, we develop a new, computationally efficient method for simultaneously mapping multiple gene expression quantitative trait loci that directly uses all of the available data. Information shared across gene expression traits is captured in a way that makes minimal assumptions about the statistical properties of the data. The method produces easy-to-interpret measures of statistical significance for both individual loci and the overall joint significance of multiple loci selected for a given expression trait. We apply the new method to a cross between two strains of the budding yeast Saccharomyces cerevisiae, and estimate that at least 37% of all gene expression traits show two simultaneous linkages, where we have allowed for epistatic interactions. Pairs of jointly linking quantitative trait loci are identified with high confidence for 170 gene expression traits, where it is expected that both loci are true positives for at least 153 traits. In addition, we are able to show that epistatic interactions contribute to gene expression variation for at least 14% of all traits. We compare the proposed approach to an exhaustive two-dimensional scan over all pairs of loci. Surprisingly, we demonstrate that an exhaustive two-dimensional scan is less powerful than the sequential search used here. In addition, we show that a two-dimensional scan does not truly allow one to test for simultaneous linkage, and the statistical significance measured from this existing method cannot be interpreted among many traits.
Figures
Similar articles
-
Barcoded bulk QTL mapping reveals highly polygenic and epistatic architecture of complex traits in yeast.Elife. 2022 Feb 11;11:e73983. doi: 10.7554/eLife.73983. Elife. 2022. PMID: 35147078 Free PMC article.
-
Genetics of single-cell protein abundance variation in large yeast populations.Nature. 2014 Feb 27;506(7489):494-7. doi: 10.1038/nature12904. Epub 2014 Jan 8. Nature. 2014. PMID: 24402228 Free PMC article.
-
Simultaneous fine mapping of closely linked epistatic quantitative trait loci using combined linkage disequilibrium and linkage with a general pedigree.Genet Sel Evol. 2008 May-Jun;40(3):265-78. doi: 10.1186/1297-9686-40-3-265. Epub 2008 Apr 10. Genet Sel Evol. 2008. PMID: 18400149 Free PMC article.
-
Genetic mapping of quantitative phenotypic traits in Saccharomyces cerevisiae.FEMS Yeast Res. 2012 Mar;12(2):215-27. doi: 10.1111/j.1567-1364.2011.00777.x. Epub 2012 Jan 24. FEMS Yeast Res. 2012. PMID: 22150948 Review.
-
Genetic dissection of complex traits in yeast: insights from studies of gene expression and other phenotypes in the BYxRM cross.Cold Spring Harb Symp Quant Biol. 2009;74:145-53. doi: 10.1101/sqb.2009.74.013. Epub 2009 Sep 4. Cold Spring Harb Symp Quant Biol. 2009. PMID: 19734204 Free PMC article. Review.
Cited by
-
eQTL Epistasis - Challenges and Computational Approaches.Front Genet. 2013 May 31;4:51. doi: 10.3389/fgene.2013.00051. eCollection 2013. Front Genet. 2013. PMID: 23755066 Free PMC article.
-
A decision-theory approach to interpretable set analysis for high-dimensional data.Biometrics. 2013 Sep;69(3):614-23. doi: 10.1111/biom.12060. Epub 2013 Aug 2. Biometrics. 2013. PMID: 23909925 Free PMC article.
-
Capturing heterogeneity in gene expression studies by surrogate variable analysis.PLoS Genet. 2007 Sep;3(9):1724-35. doi: 10.1371/journal.pgen.0030161. Epub 2007 Aug 1. PLoS Genet. 2007. PMID: 17907809 Free PMC article.
-
Gene expression profiling reveals early cellular responses to intracellular magnetic labeling with superparamagnetic iron oxide nanoparticles.Magn Reson Med. 2010 Apr;63(4):1031-43. doi: 10.1002/mrm.22290. Magn Reson Med. 2010. PMID: 20373404 Free PMC article.
-
Systems genetics, bioinformatics and eQTL mapping.Genetica. 2010 Oct;138(9-10):915-24. doi: 10.1007/s10709-010-9480-x. Epub 2010 Sep 3. Genetica. 2010. PMID: 20811929 Review.
References
-
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. - PubMed
-
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotech. 1996;14:1675–1680. - PubMed
-
- MacBeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. - PubMed
-
- Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
