

Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes
- PMID: 16024819
- PMCID: PMC1182216
- DOI: 10.1101/gr.3715005
Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes
Abstract
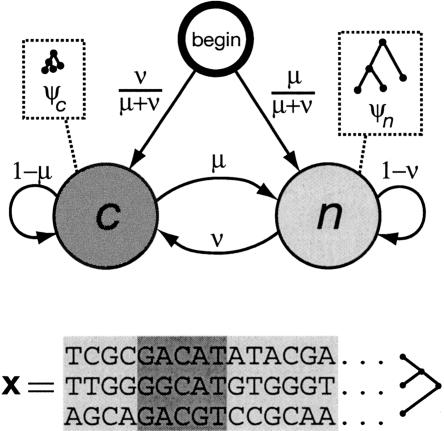
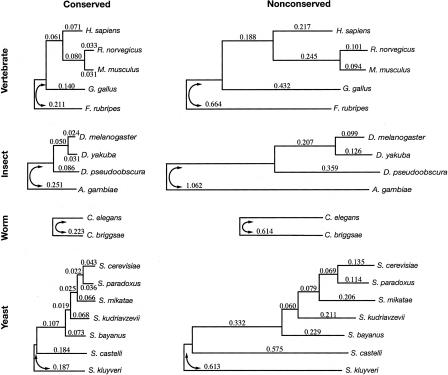
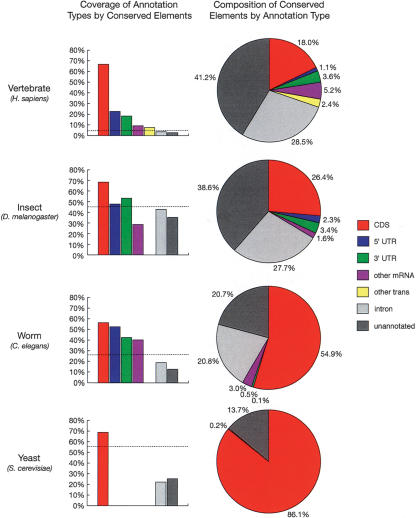
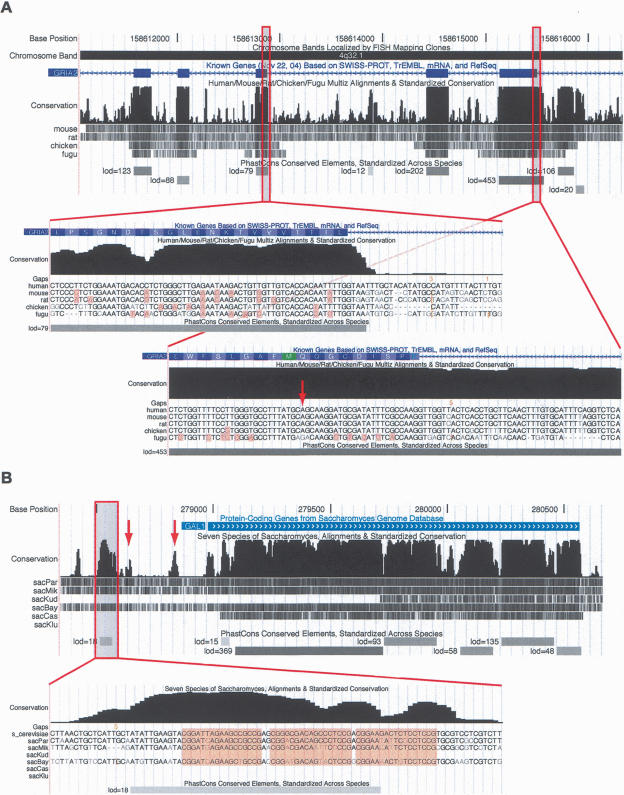
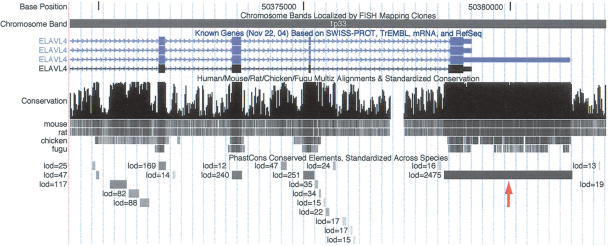
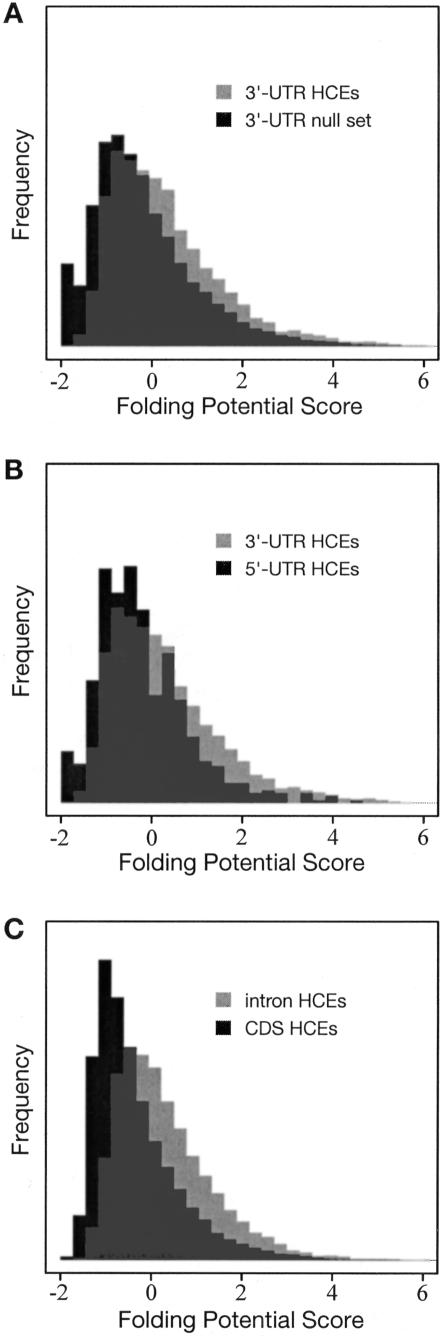
We have conducted a comprehensive search for conserved elements in vertebrate genomes, using genome-wide multiple alignments of five vertebrate species (human, mouse, rat, chicken, and Fugu rubripes). Parallel searches have been performed with multiple alignments of four insect species (three species of Drosophila and Anopheles gambiae), two species of Caenorhabditis, and seven species of Saccharomyces. Conserved elements were identified with a computer program called phastCons, which is based on a two-state phylogenetic hidden Markov model (phylo-HMM). PhastCons works by fitting a phylo-HMM to the data by maximum likelihood, subject to constraints designed to calibrate the model across species groups, and then predicting conserved elements based on this model. The predicted elements cover roughly 3%-8% of the human genome (depending on the details of the calibration procedure) and substantially higher fractions of the more compact Drosophila melanogaster (37%-53%), Caenorhabditis elegans (18%-37%), and Saccharaomyces cerevisiae (47%-68%) genomes. From yeasts to vertebrates, in order of increasing genome size and general biological complexity, increasing fractions of conserved bases are found to lie outside of the exons of known protein-coding genes. In all groups, the most highly conserved elements (HCEs), by log-odds score, are hundreds or thousands of bases long. These elements share certain properties with ultraconserved elements, but they tend to be longer and less perfectly conserved, and they overlap genes of somewhat different functional categories. In vertebrates, HCEs are associated with the 3' UTRs of regulatory genes, stable gene deserts, and megabase-sized regions rich in moderately conserved noncoding sequences. Noncoding HCEs also show strong statistical evidence of an enrichment for RNA secondary structure.
Figures
References
-
- Bejerano, G., Haussler, D., and Blanchette, M. 2004a. Into the heart of darkness: Large-scale clustering of human non-coding DNA. Bioinformatics 20: I40–I48. - PubMed
-
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W., Mattick, J., and Haussler, D. 2004b. Ultraconserved elements in the human genome. Science 304: 1321–1325. - PubMed
-
- Bergman, C.M. and Kreitman, M. 2001. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res. 11: 1335–1345. - PubMed
Web site references
-
- http://www.cse.ucsc.edu/~acs/conservation; Supplemental data for this study.
-
- http://genome.ucsc.edu; UC Santa Cruz Genome Browser. - PubMed
-
- http://genome.ucsc.edu/cgi-bin/hgTables; UC Santa Cruz Table Browser.
-
- http://www.genetics.wustl.edu/saccharomycesgenomes/Contigs; download page for yeast sequence data, Washington University, St. Louis.
-
- http://www.broad.mit.edu/ftp/pub/annotation/fungi/comp_yeasts; download page for yeast sequence data, Broad Institute.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases