

Immune responses and disease enhancement during respiratory syncytial virus infection
- PMID: 16020689
- PMCID: PMC1195968
- DOI: 10.1128/CMR.18.3.541-555.2005
Immune responses and disease enhancement during respiratory syncytial virus infection
Abstract
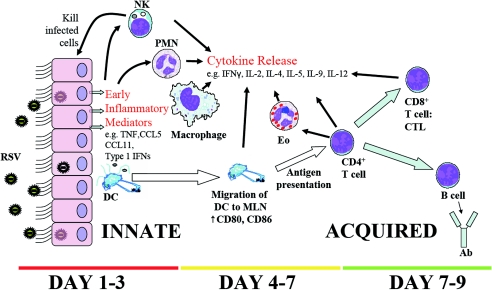
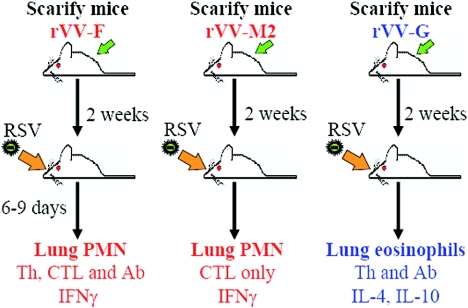
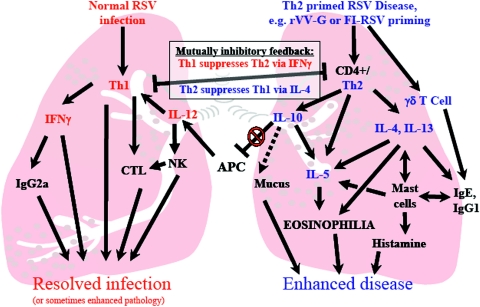
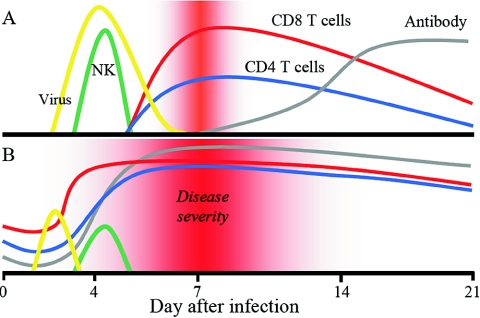
Respiratory syncytial virus (RSV) is one of the commonest and most troublesome viruses of infancy. It causes most cases of bronchiolitis, which is associated with wheezing in later childhood. In primary infection, the peak of disease typically coincides with the development of specific T- and B-cell responses, which seem, in large part, to be responsible for disease. Animal models clearly show that a range of immune responses can enhance disease severity, particularly after vaccination with formalin-inactivated RSV. Prior immune sensitization leads to exuberant chemokine production, an excessive cellular influx, and an overabundance of cytokines during RSV challenge. Under different circumstances, specific mediators and T-cell subsets and antibody-antigen immune complex deposition are incriminated as major factors in disease. Animal models of immune enhancement permit a deep understanding of the role of specific immune responses in RSV disease, assist in vaccine design, and indicate which immunomodulatory therapy might be beneficial to children with bronchiolitis.
Figures

Similar articles
-
Antiviral immune responses and lung inflammation after respiratory syncytial virus infection.Proc Am Thorac Soc. 2005;2(2):121-5. doi: 10.1513/pats.200504-032AW. Proc Am Thorac Soc. 2005. PMID: 16113479 Review.
-
Immunopathology of RSV infection: prospects for developing vaccines without this complication.Rev Med Virol. 2007 Jan-Feb;17(1):5-34. doi: 10.1002/rmv.518. Rev Med Virol. 2007. PMID: 17004293 Review.
-
Anti-G protein antibody responses to respiratory syncytial virus infection or vaccination are associated with inhibition of G protein CX3C-CX3CR1 binding and leukocyte chemotaxis.J Infect Dis. 2004 Dec 1;190(11):1936-40. doi: 10.1086/425516. Epub 2004 Oct 28. J Infect Dis. 2004. PMID: 15529257 Clinical Trial.
-
Advances in and the potential of vaccines for respiratory syncytial virus.Expert Rev Respir Med. 2013 Aug;7(4):411-27. doi: 10.1586/17476348.2013.814409. Expert Rev Respir Med. 2013. PMID: 23964629 Review.
-
Clinical and epidemiologic features of respiratory syncytial virus.Curr Top Microbiol Immunol. 2013;372:39-57. doi: 10.1007/978-3-642-38919-1_2. Curr Top Microbiol Immunol. 2013. PMID: 24362683
Cited by
-
Sirtuin 1 Regulates Dendritic Cell Activation and Autophagy during Respiratory Syncytial Virus-Induced Immune Responses.J Immunol. 2015 Aug 15;195(4):1637-46. doi: 10.4049/jimmunol.1500326. Epub 2015 Jul 8. J Immunol. 2015. PMID: 26157176 Free PMC article.
-
Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease.Front Immunol. 2019 Mar 22;10:548. doi: 10.3389/fimmu.2019.00548. eCollection 2019. Front Immunol. 2019. PMID: 30967872 Free PMC article. Review.
-
Natural killer cell NKG2D and granzyme B are critical for allergic pulmonary inflammation.J Allergy Clin Immunol. 2014 Mar;133(3):827-35.e3. doi: 10.1016/j.jaci.2013.09.048. Epub 2013 Nov 28. J Allergy Clin Immunol. 2014. PMID: 24290277 Free PMC article.
-
Liver Dysfunction in Severe Sepsis from Respiratory Syncytial Virus.J Pediatr Intensive Care. 2018 Jun;7(2):110-114. doi: 10.1055/s-0037-1612609. Epub 2017 Dec 21. J Pediatr Intensive Care. 2018. PMID: 31073482 Free PMC article.
-
Current concepts and progress in RSV vaccine development.Expert Rev Vaccines. 2014 Mar;13(3):333-44. doi: 10.1586/14760584.2014.878653. Epub 2014 Jan 9. Expert Rev Vaccines. 2014. PMID: 24405366 Free PMC article. Review.
References
-
- Abdallah, A., K. E. Rowland, S. K. Schepetiuk, L. B. To, and P. Bardy. 2003. An outbreak of respiratory syncytial virus infection in a bone marrow transplant unit: effect on engraftment and outcome of pneumonia without specific antiviral treatment. Bone Marrow Transplant. 32:195-203. - PubMed
-
- Aberle, J. H., S. W. Aberle, M. N. Dworzak, C. W. Mandl, W. Rebhandl, G. Vollnhofer, M. Kundi, and K. T. Popow. 1999. Reduced interferon-gamma expression in peripheral blood mononuclear cells of infants with severe respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 160:1263-1268. - PubMed
-
- Adkins, B., C. LeClerc, and S. Marshall-Clarke. 2004. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4:553-564. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
