

Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus
- PMID: 15781584
- PMCID: PMC2213100
- DOI: 10.1084/jem.20041901
Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus
Abstract
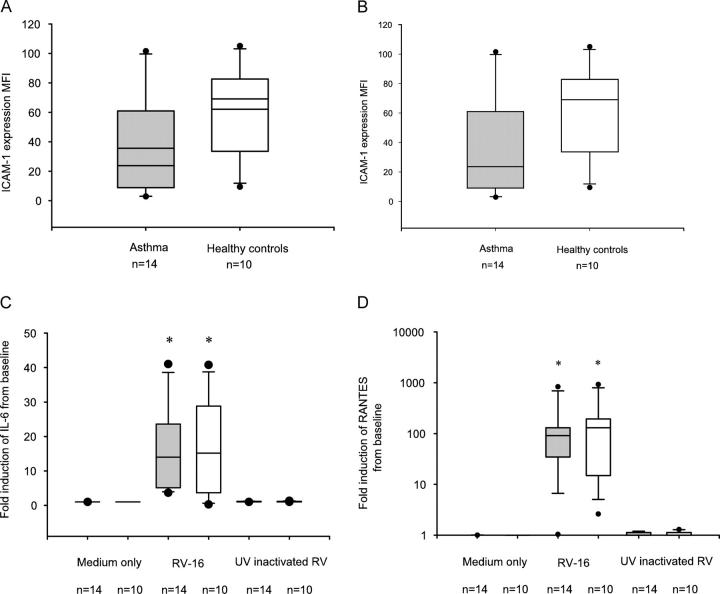
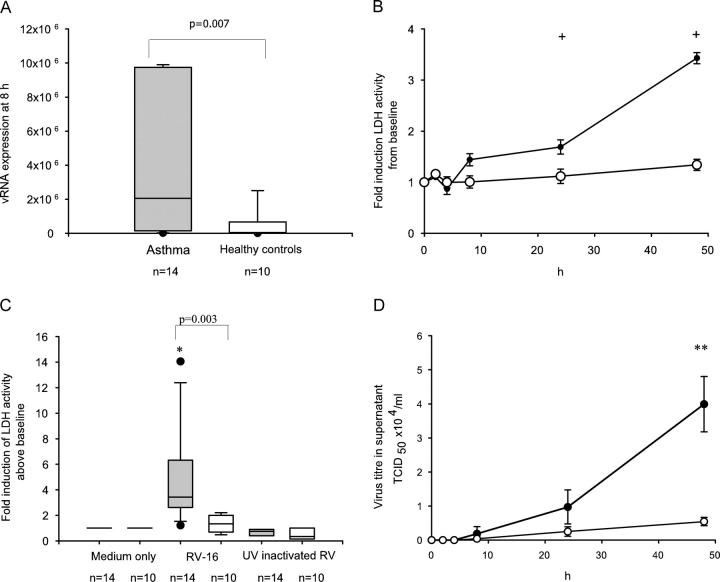
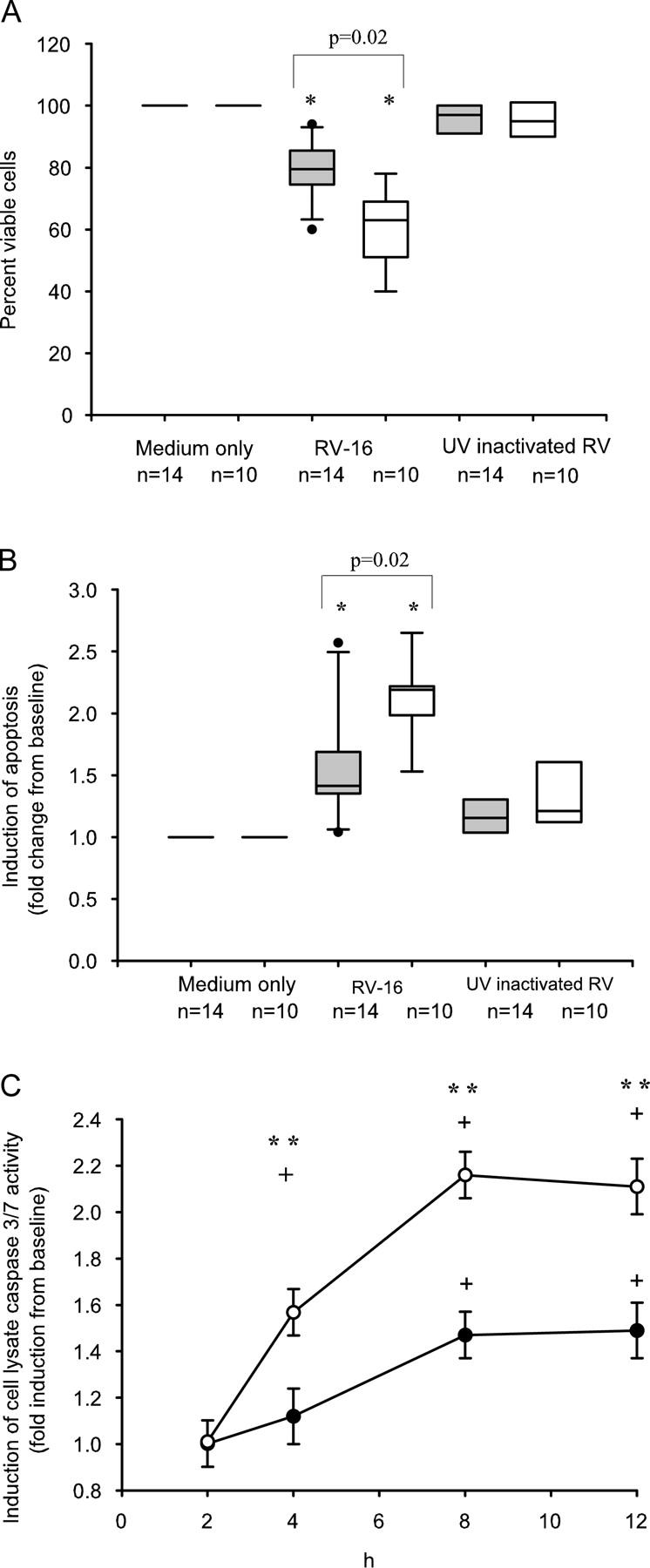
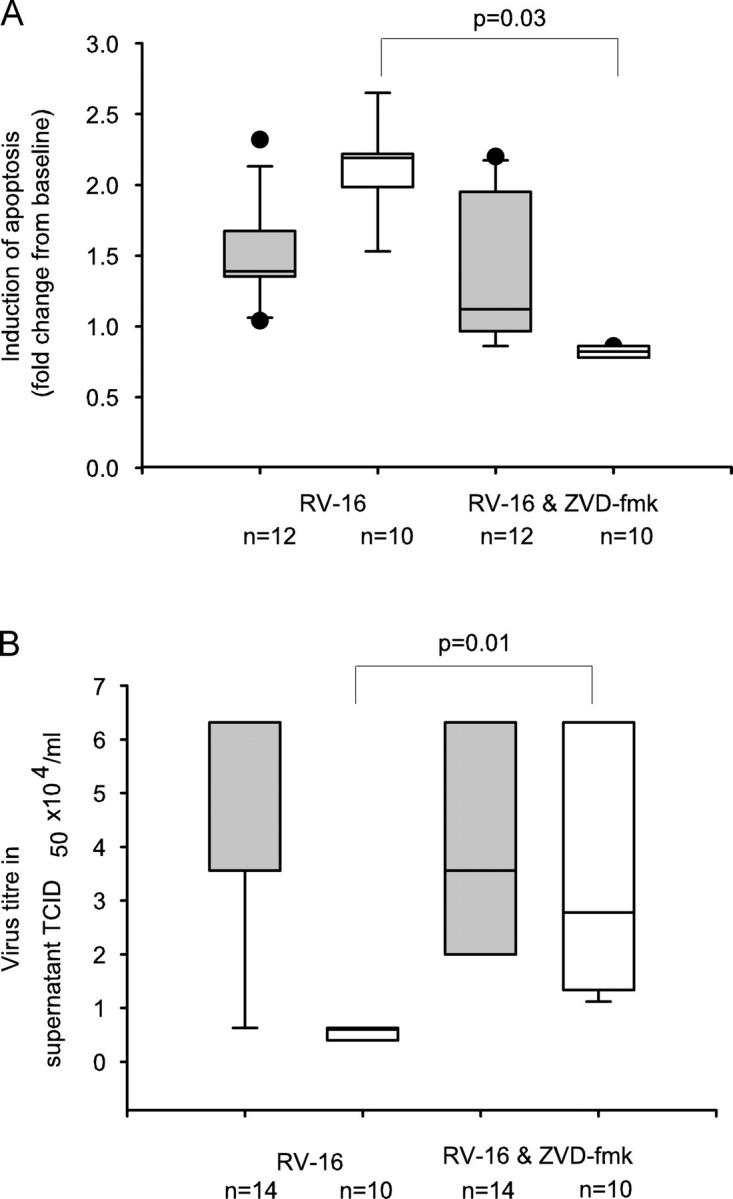
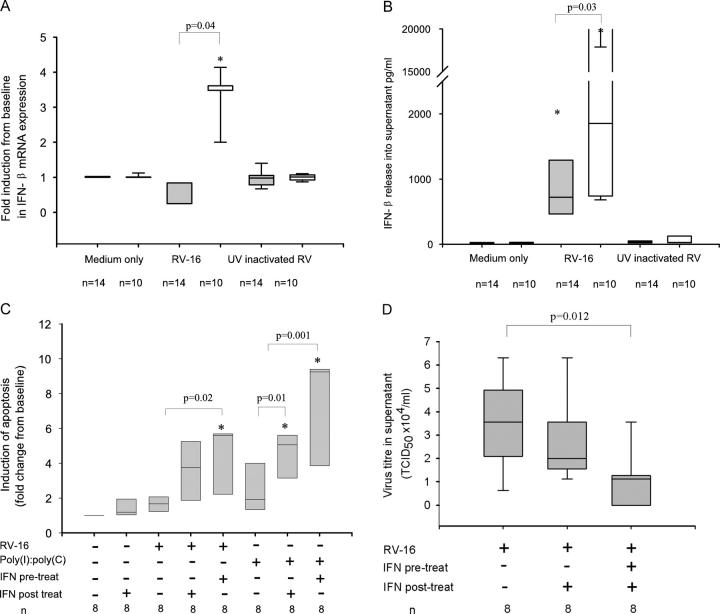
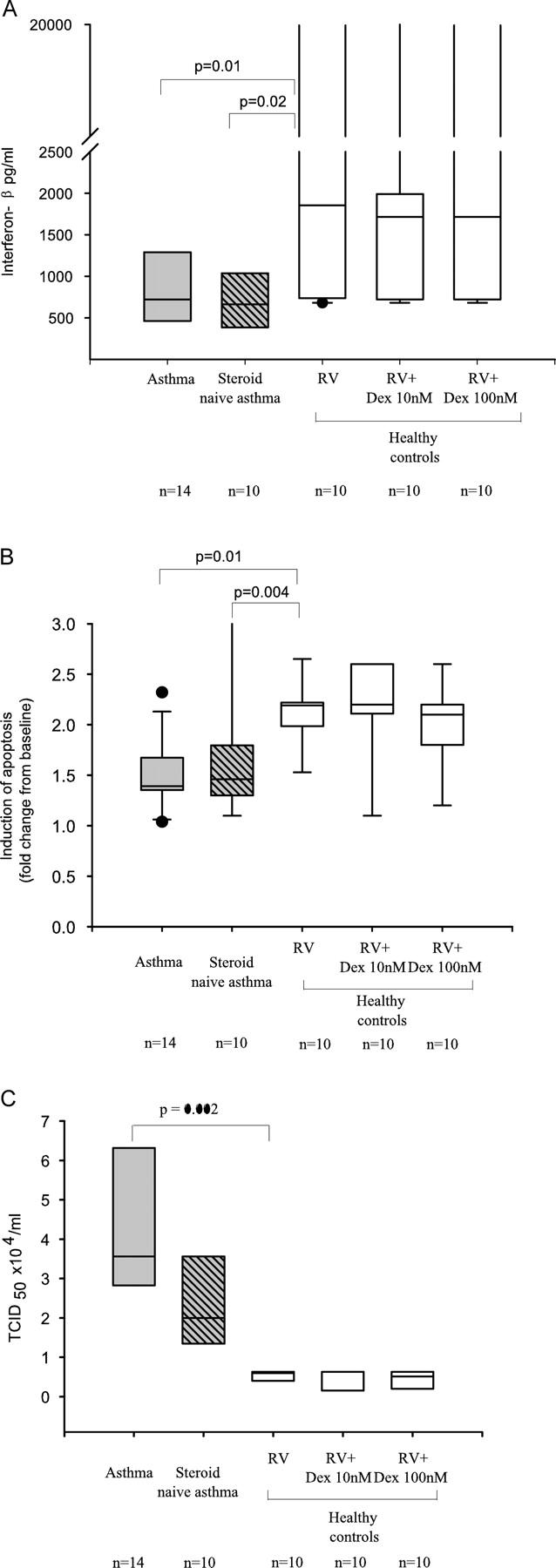
Rhinoviruses are the major trigger of acute asthma exacerbations and asthmatic subjects are more susceptible to these infections. To investigate the underlying mechanisms of this increased susceptibility, we examined virus replication and innate responses to rhinovirus (RV)-16 infection of primary bronchial epithelial cells from asthmatic and healthy control subjects. Viral RNA expression and late virus release into supernatant was increased 50- and 7-fold, respectively in asthmatic cells compared with healthy controls. Virus infection induced late cell lysis in asthmatic cells but not in normal cells. Examination of the early cellular response to infection revealed impairment of virus induced caspase 3/7 activity and of apoptotic responses in the asthmatic cultures. Inhibition of apoptosis in normal cultures resulted in enhanced viral yield, comparable to that seen in infected asthmatic cultures. Examination of early innate immune responses revealed profound impairment of virus-induced interferon-beta mRNA expression in asthmatic cultures and they produced >2.5 times less interferon-beta protein. In infected asthmatic cells, exogenous interferon-beta induced apoptosis and reduced virus replication, demonstrating a causal link between deficient interferon-beta, impaired apoptosis and increased virus replication. These data suggest a novel use for type I interferons in the treatment or prevention of virus-induced asthma exacerbations.
Figures
Similar articles
-
Exogenous IFN-β has antiviral and anti-inflammatory properties in primary bronchial epithelial cells from asthmatic subjects exposed to rhinovirus.J Allergy Clin Immunol. 2011 May;127(5):1148-54.e9. doi: 10.1016/j.jaci.2011.01.023. Epub 2011 Feb 16. J Allergy Clin Immunol. 2011. PMID: 21329968
-
Diversity in the bronchial epithelial cell response to infection with different rhinovirus strains.Respirology. 2009 Mar;14(2):180-6. doi: 10.1111/j.1440-1843.2009.01480.x. Epub 2009 Feb 4. Respirology. 2009. PMID: 19207121
-
Impaired type I and type III interferon induction and rhinovirus control in human cystic fibrosis airway epithelial cells.Thorax. 2012 Jun;67(6):517-25. doi: 10.1136/thoraxjnl-2011-200405. Epub 2012 Jan 2. Thorax. 2012. Retraction in: Thorax. 2013 Sep;68(9):886. doi: 10.1136/thoraxjnl-2011-200405ret PMID: 22213737 Retracted.
-
Innate immunity in the pathogenesis of virus-induced asthma exacerbations.Proc Am Thorac Soc. 2007 Jul;4(3):267-70. doi: 10.1513/pats.200701-030AW. Proc Am Thorac Soc. 2007. PMID: 17607011 Review.
-
Regulation and Function of Interferon-Lambda (IFNλ) and Its Receptor in Asthma.Front Immunol. 2021 Nov 10;12:731807. doi: 10.3389/fimmu.2021.731807. eCollection 2021. Front Immunol. 2021. PMID: 34899691 Free PMC article. Review.
Cited by
-
Viruses and asthma: the role of common respiratory viruses in asthma and its potential meaning for SARS-CoV-2.Immunology. 2020 Oct;161(2):83-93. doi: 10.1111/imm.13240. Epub 2020 Aug 17. Immunology. 2020. PMID: 32687609 Free PMC article. Review.
-
Distinct spatial and temporal roles for Th1, Th2, and Th17 cells in asthma.Front Immunol. 2022 Aug 12;13:974066. doi: 10.3389/fimmu.2022.974066. eCollection 2022. Front Immunol. 2022. PMID: 36032162 Free PMC article. Review.
-
Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons.J Allergy Clin Immunol. 2015 Jul;136(1):177-188.e11. doi: 10.1016/j.jaci.2014.11.039. Epub 2015 Jan 25. J Allergy Clin Immunol. 2015. PMID: 25630941 Free PMC article.
-
Budesonide and formoterol reduce early innate anti-viral immune responses in vitro.PLoS One. 2011;6(11):e27898. doi: 10.1371/journal.pone.0027898. Epub 2011 Nov 18. PLoS One. 2011. PMID: 22125636 Free PMC article.
-
Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response.PLoS One. 2012;7(9):e44580. doi: 10.1371/journal.pone.0044580. Epub 2012 Sep 6. PLoS One. 2012. PMID: 22970254 Free PMC article.
References
-
- Johnston, S.L., P.K. Pattemore, G. Sanderson, S. Smith, M.J. Campbell, L.K. Josephs, A. Cunningham, B.S. Robinson, S.H. Myint, M.E. Ward, et al. 1996. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am. J. Respir. Crit. Care Med. 154:654–660. - PubMed
-
- Reddel, H., S. Ware, G. Marks, C. Salome, C. Jenkins, and A. Woolcock. 1999. Differences between asthma exacerbations and poor asthma control. Lancet. 353:364–369. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
