

Comparative sequencing provides insights about the structure and conservation of marsupial and monotreme genomes
- PMID: 15718282
- PMCID: PMC549084
- DOI: 10.1073/pnas.0408539102
Comparative sequencing provides insights about the structure and conservation of marsupial and monotreme genomes
Abstract
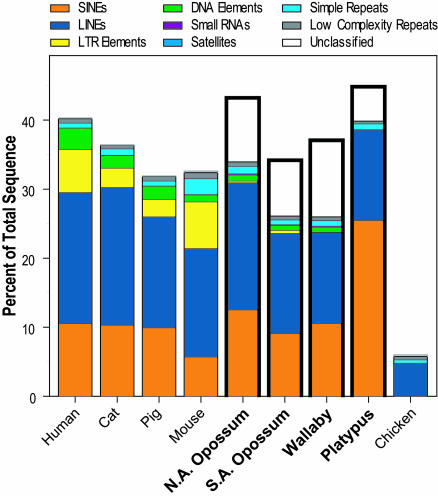
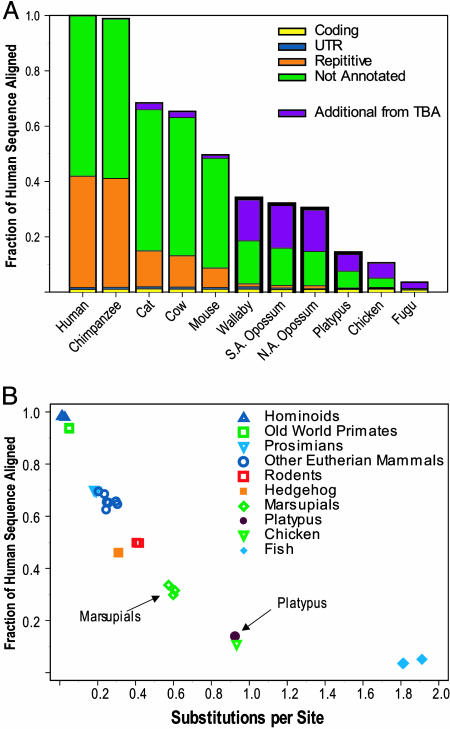
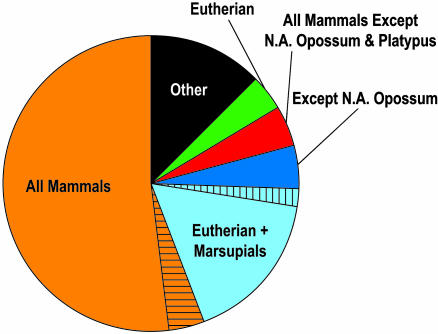
Sequencing and comparative analyses of genomes from multiple vertebrates are providing insights about the genetic basis for biological diversity. To date, these efforts largely have focused on eutherian mammals, chicken, and fish. In this article, we describe the generation and study of genomic sequences from noneutherian mammals, a group of species occupying unusual phylogenetic positions. A large sequence data set (totaling >5 Mb) was generated for the same orthologous region in three marsupial (North American opossum, South American opossum, and Australian tammar wallaby) and one monotreme (platypus) genomes. These ancient mammalian genomes are characterized by unusual architectural features with respect to G + C and repeat content, as well as compression relative to human. Approximately 14% and 34% of the human sequence forms alignments with the orthologous sequence from platypus and the marsupials, respectively; these numbers are distinctly lower than that observed with nonprimate eutherian mammals (45-70%). The alignable sequences between human and each marsupial species are not completely overlapping (only 80% common to all three species) nor are the platypus-alignable sequences completely contained within the marsupial-alignable sequences. Phylogenetic analysis of synonymous coding positions reveals that platypus has a notably long branch length, with the human-platypus substitution rate being on average 55% greater than that seen with human-marsupial pairs. Finally, analyses of the major mammalian lineages reveal distinct patterns with respect to the common presence of evolutionarily conserved vertebrate sequences. Our results confirm that genomic sequence from noneutherian mammals can contribute uniquely to unraveling the functional and evolutionary histories of the mammalian genome.
Figures

Similar articles
-
Characterisation of monotreme caseins reveals lineage-specific expansion of an ancestral casein locus in mammals.Reprod Fertil Dev. 2009;21(8):1015-27. doi: 10.1071/RD09083. Reprod Fertil Dev. 2009. PMID: 19874726
-
Immunome database for marsupials and monotremes.BMC Immunol. 2011 Aug 19;12:48. doi: 10.1186/1471-2172-12-48. BMC Immunol. 2011. PMID: 21854560 Free PMC article.
-
The evolution of marsupial and monotreme chromosomes.Cytogenet Genome Res. 2012;137(2-4):113-29. doi: 10.1159/000339433. Epub 2012 Jul 6. Cytogenet Genome Res. 2012. PMID: 22777195 Review.
-
Distribution of keratin and associated proteins in the epidermis of monotreme, marsupial, and placental mammals.J Morphol. 2003 Oct;258(1):49-66. doi: 10.1002/jmor.10118. J Morphol. 2003. PMID: 12905534
-
Marsupial and monotreme genomes.Genome Dyn. 2006;2:111-122. doi: 10.1159/000095099. Genome Dyn. 2006. PMID: 18753774 Review.
Cited by
-
Effort required to finish shotgun-generated genome sequences differs significantly among vertebrates.BMC Genomics. 2010 Jan 11;11:21. doi: 10.1186/1471-2164-11-21. BMC Genomics. 2010. PMID: 20064230 Free PMC article.
-
A body map of super-enhancers and their function in pig.Front Vet Sci. 2023 Oct 6;10:1239965. doi: 10.3389/fvets.2023.1239965. eCollection 2023. Front Vet Sci. 2023. PMID: 37869495 Free PMC article.
-
Biased distributions and decay of long interspersed nuclear elements in the chicken genome.Genetics. 2008 Jan;178(1):573-81. doi: 10.1534/genetics.106.061861. Epub 2007 Oct 18. Genetics. 2008. PMID: 17947446 Free PMC article.
-
A reference genome for the critically endangered woylie, Bettongia penicillata ogilbyi.GigaByte. 2021 Dec 10;2021:gigabyte35. doi: 10.46471/gigabyte.35. eCollection 2021. GigaByte. 2021. PMID: 36824341 Free PMC article.
-
Evolution of the "OR37" subfamily of olfactory receptors: a cross-species comparison.J Mol Evol. 2006 Apr;62(4):460-72. doi: 10.1007/s00239-005-0093-4. Epub 2006 Mar 17. J Mol Evol. 2006. PMID: 16547640
References
-
- Cooper, G. M. & Sidow, A. (2003) Curr. Opin. Genet. Dev. 13, 604-610. - PubMed
-
- Ureta-Vidal, A., Ettwiller, L. & Birney, E. (2003) Nat. Rev. Genet. 4, 251-262. - PubMed
-
- Aparicio, S., Chapman, J., Stupka, E., Putnam, N., Chia, J.-M., Dehal, P., Christoffels, A., Rash, S., Hoon, S., Smit, A., et al. (2002) Science 297, 1301-1310. - PubMed
-
- International Mouse Genome Sequencing Consortium (2002) Nature 420, 520-562. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
