

Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression
- PMID: 15681825
- PMCID: PMC1237049
- DOI: 10.1016/s0002-9440(10)62264-x
Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression
Abstract
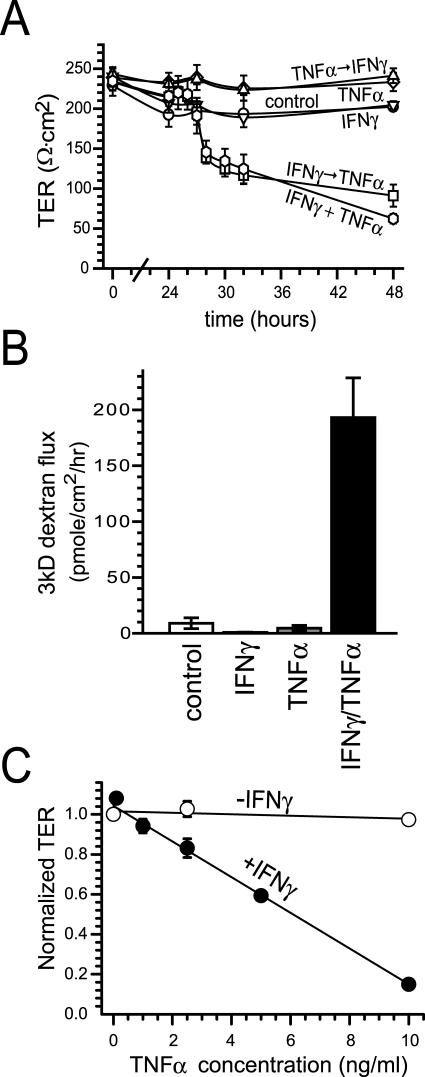
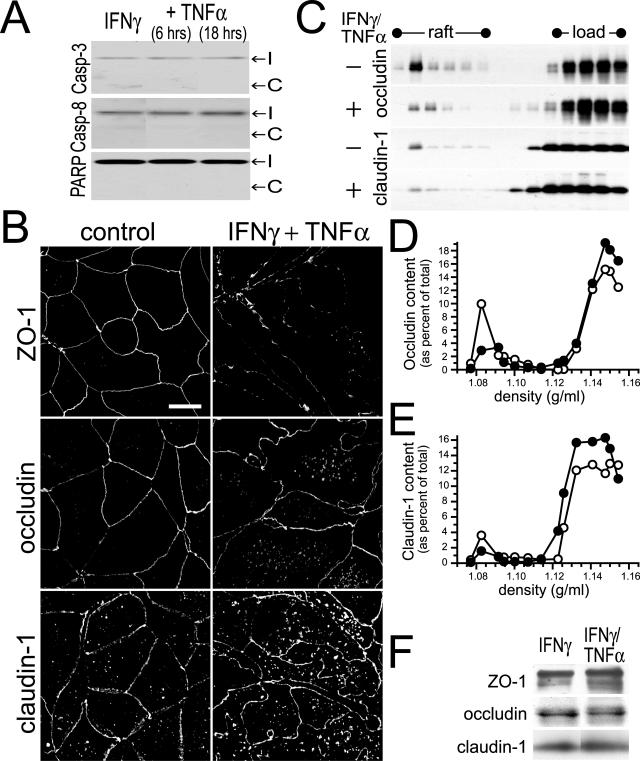
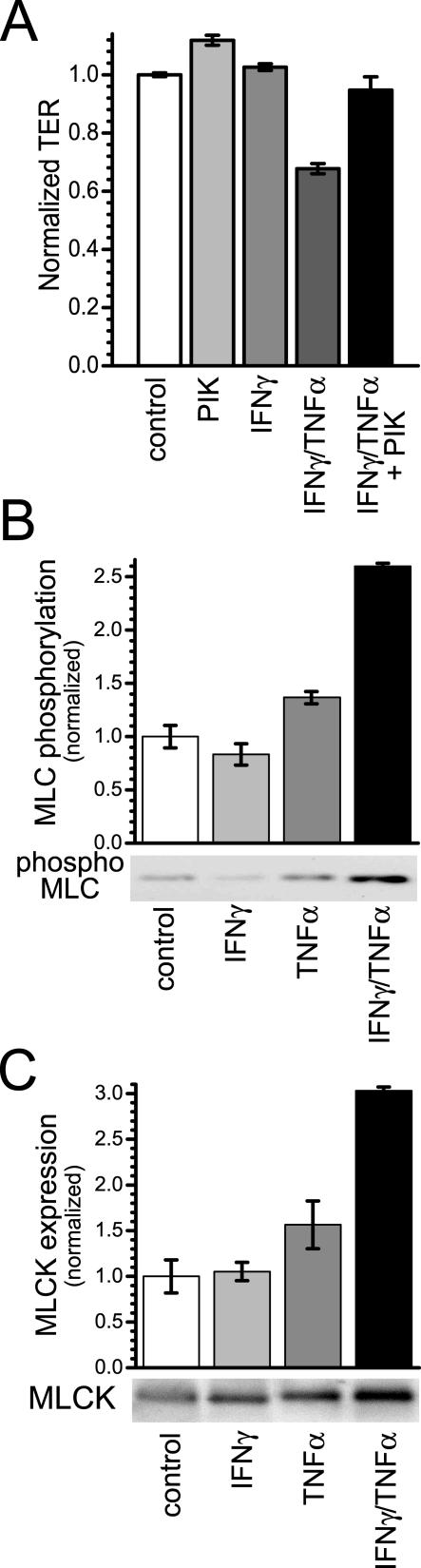
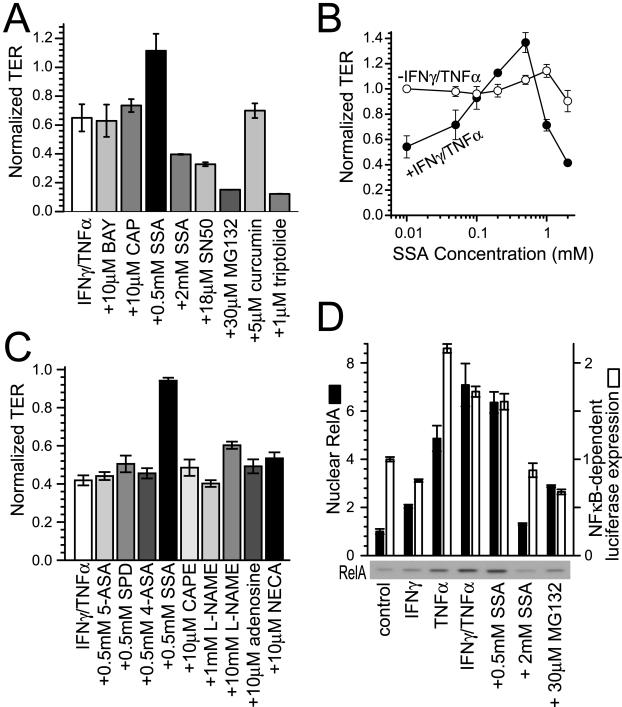
Numerous intestinal diseases are characterized by immune cell activation and compromised epithelial barrier function. We have shown that cytokine treatment of epithelial monolayers increases myosin II regulatory light chain (MLC) phosphorylation and decreases barrier function and that these are both reversed by MLC kinase (MLCK) inhibition. The aim of this study was to determine the mechanisms by which interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha regulate MLC phosphorylation and disrupt epithelial barrier function. We developed a model in which both cytokines were required for barrier dysfunction. Barrier dysfunction was also induced by TNF-alpha addition to IFN-gamma-primed, but not control, Caco-2 monolayers. TNF-alpha treatment of IFN-gamma-primed monolayers caused increases in both MLCK expression and MLC phosphorylation, suggesting that MLCK is a TNF-alpha-inducible protein. These effects of TNF-alpha were not mediated by nuclear factor-kappaB. However, at doses below those needed for nuclear factor-kappaB inhibition, sulfasalazine was able to prevent TNF-alpha-induced barrier dysfunction, MLCK up-regulation, and MLC phosphorylation. Low-dose sulfasalazine also prevented morphologically evident tight junction disruption induced by TNF-alpha. These data show that IFN-gamma can prime intestinal epithelial monolayers to respond to TNF-alpha by disrupting tight junction morphology and barrier function via MLCK up-regulation and MLC phosphorylation. These TNF-alpha-induced events can be prevented by the clinically relevant drug sulfasalazine.
Figures

Similar articles
-
Amelioration of IFN-γ and TNF-α-induced intestinal epithelial barrier dysfunction by berberine via suppression of MLCK-MLC phosphorylation signaling pathway.PLoS One. 2013 May 3;8(5):e61944. doi: 10.1371/journal.pone.0061944. Print 2013. PLoS One. 2013. PMID: 23671580 Free PMC article.
-
IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction.Gastroenterology. 2006 Oct;131(4):1153-63. doi: 10.1053/j.gastro.2006.08.022. Epub 2006 Aug 22. Gastroenterology. 2006. PMID: 17030185 Free PMC article.
-
Blockade of hypoxia-inducible factor-1α by YC-1 attenuates interferon-γ and tumor necrosis factor-α-induced intestinal epithelial barrier dysfunction.Cytokine. 2011 Dec;56(3):581-8. doi: 10.1016/j.cyto.2011.08.023. Epub 2011 Sep 3. Cytokine. 2011. PMID: 21890376
-
The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases.Int J Mol Sci. 2020 May 18;21(10):3550. doi: 10.3390/ijms21103550. Int J Mol Sci. 2020. PMID: 32443411 Free PMC article. Review.
-
Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis.Int J Mol Sci. 2020 Feb 3;21(3):993. doi: 10.3390/ijms21030993. Int J Mol Sci. 2020. PMID: 32028590 Free PMC article. Review.
Cited by
-
Rapid Evaluation of Intestinal Paracellular Permeability Using the Human Enterocytic-Like Caco-2/TC7 Cell Line.Methods Mol Biol. 2021;2367:13-26. doi: 10.1007/7651_2021_366. Methods Mol Biol. 2021. PMID: 33730353
-
Mechanisms and functional implications of intestinal barrier defects.Dig Dis. 2009;27(4):443-9. doi: 10.1159/000233282. Epub 2009 Nov 4. Dig Dis. 2009. PMID: 19897958 Free PMC article. Review.
-
The role of mir-197-3p in regulating the tight junction permeability of celiac disease patients under gluten free diet.Mol Biol Rep. 2023 Mar;50(3):2007-2014. doi: 10.1007/s11033-022-08147-w. Epub 2022 Dec 19. Mol Biol Rep. 2023. PMID: 36536183
-
Dietary pterostilbene supplementation attenuates intestinal damage and immunological stress of broiler chickens challenged with lipopolysaccharide.J Anim Sci. 2020 Jan 1;98(1):skz373. doi: 10.1093/jas/skz373. J Anim Sci. 2020. PMID: 31822918 Free PMC article.
-
Enhanced gastrointestinal passive paracellular permeability contributes to the obesity-associated hyperoxaluria.Am J Physiol Gastrointest Liver Physiol. 2019 Jan 1;316(1):G1-G14. doi: 10.1152/ajpgi.00266.2018. Epub 2018 Oct 11. Am J Physiol Gastrointest Liver Physiol. 2019. PMID: 30307745 Free PMC article.
References
-
- Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–291. - PubMed
-
- Irvine EJ, Marshall JK. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology. 2000;119:1740–1744. - PubMed
-
- Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–1439. - PubMed
-
- Yacyshyn BR, Meddings JB. CD45RO expression on circulating CD19+ B cells in Crohn’s disease correlates with intestinal permeability. Gastroenterology. 1995;108:132–137. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
