

Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth
- PMID: 15657416
- PMCID: PMC544001
- DOI: 10.1128/MCB.25.3.865-878.2005
Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth
Abstract
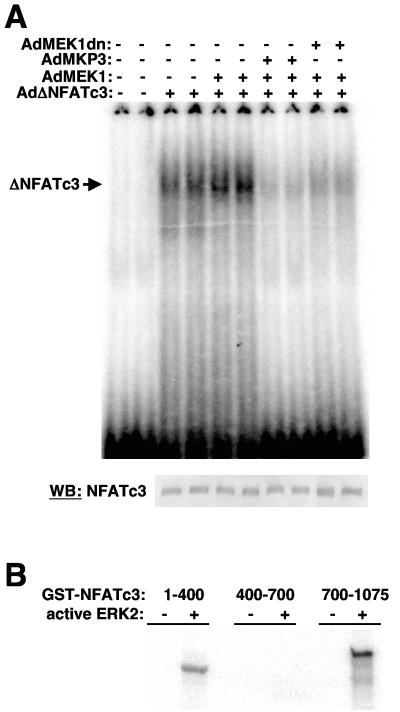
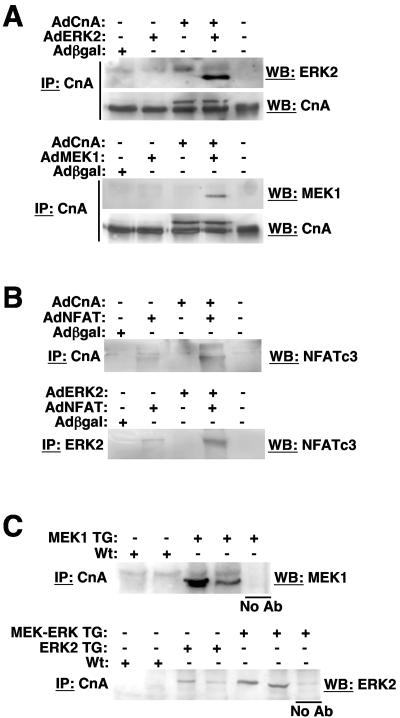
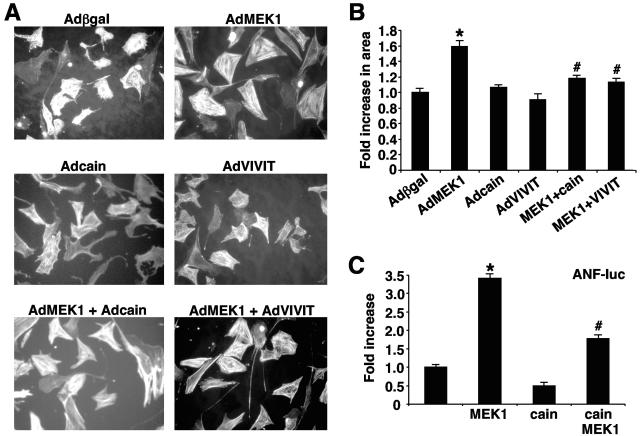
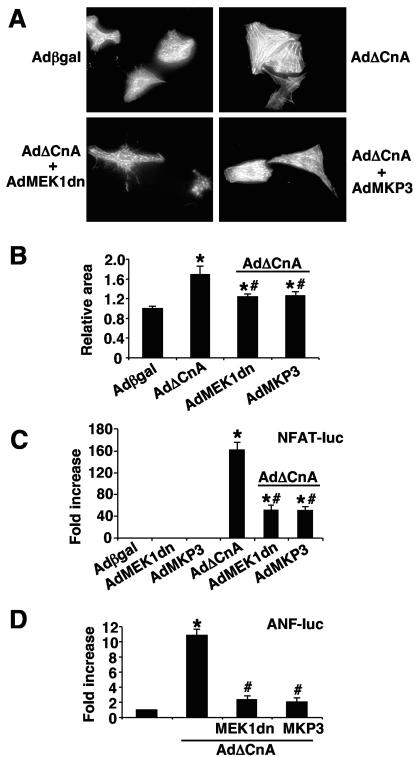
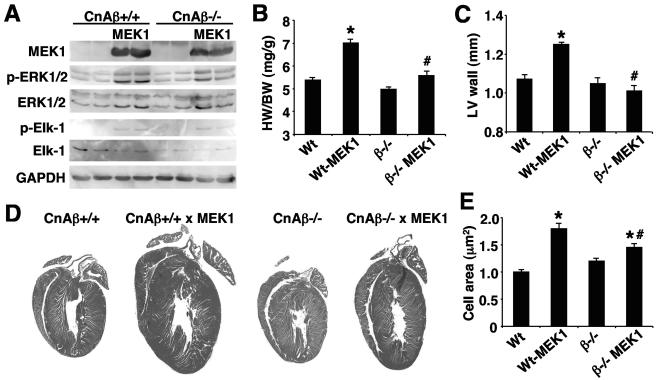
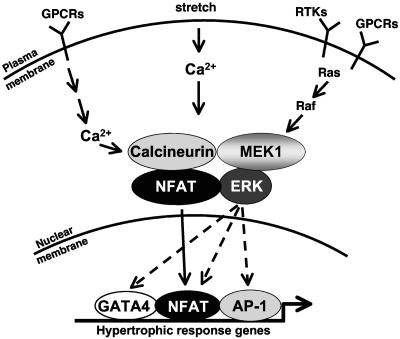
MEK1, a member of the mitogen-activated protein kinase (MAPK) cascade that directly activates extracellular signal-regulated kinase (ERK), induces cardiac hypertrophy in transgenic mice. Calcineurin is a calcium-regulated protein phosphatase that also functions as a positive regulator of cardiac hypertrophic growth through a direct mechanism involving activation of nuclear factor of activated T-cell (NFAT) transcription factors. Here we determined that calcineurin-NFAT and MEK1-ERK1/2 signaling pathways are interdependent in cardiomyocytes, where they directly coregulate the hypertrophic growth response. For example, genetic deletion of the calcineurin Abeta gene reduced the hypertrophic response elicited by an activated MEK1 transgene in the heart, while inhibition of calcineurin or NFAT in cultured neonatal cardiomyocytes also blunted the hypertrophic response driven by activated MEK1. Conversely, targeted inhibition of MEK1-ERK1/2 signaling in cultured cardiomyocytes attenuated the hypertrophic growth response directed by activated calcineurin. However, targeted inhibition of MEK1-ERK1/2 signaling did not directly affect calcineurin-NFAT activation, nor was MEK1-ERK1/2 activation altered by targeted inhibition of calcineurin-NFAT. Mechanistically, we show that MEK1-ERK1/2 signaling augments NFAT transcriptional activity independent of calcineurin, independent of changes in NFAT nuclear localization, and independent of alterations in NFAT transactivation potential. In contrast, MEK1-ERK1/2 signaling enhances NFAT-dependent gene expression through an indirect mechanism involving induction of cardiac AP-1 activity, which functions as a necessary NFAT-interacting partner. As a second mechanism, MEK1-ERK1/2 and calcineurin-NFAT proteins form a complex in cardiac myocytes, resulting in direct phosphorylation of NFATc3 within its C terminus. MEK1-ERK1/2-mediated phosphorylation of NFATc3 directly augmented its DNA binding activity, while inhibition of MEK1-ERK1/2 signaling reduced NFATc3 DNA binding activity. Collectively, these results indicate that calcineurin-NFAT and MEK1-ERK1/2 pathways constitute a codependent signaling module in cardiomyocytes that coordinately regulates the growth response through two distinct mechanisms.
Figures

Similar articles
-
Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs.Cardiovasc Res. 2004 Aug 15;63(3):467-75. doi: 10.1016/j.cardiores.2004.01.021. Cardiovasc Res. 2004. PMID: 15276472 Review.
-
The DnaJ-related factor Mrj interacts with nuclear factor of activated T cells c3 and mediates transcriptional repression through class II histone deacetylase recruitment.Mol Cell Biol. 2005 Nov;25(22):9936-48. doi: 10.1128/MCB.25.22.9936-9948.2005. Mol Cell Biol. 2005. PMID: 16260608 Free PMC article.
-
Interference of antihypertrophic molecules and signaling pathways with the Ca2+-calcineurin-NFAT cascade in cardiac myocytes.Cardiovasc Res. 2004 Aug 15;63(3):450-7. doi: 10.1016/j.cardiores.2004.04.002. Cardiovasc Res. 2004. PMID: 15276470 Review.
-
Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling.J Clin Invest. 2003 May;111(10):1475-86. doi: 10.1172/JCI17295. J Clin Invest. 2003. PMID: 12750397 Free PMC article.
-
Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes.Proc Natl Acad Sci U S A. 2002 Aug 20;99(17):11363-8. doi: 10.1073/pnas.162100799. Epub 2002 Aug 12. Proc Natl Acad Sci U S A. 2002. PMID: 12177418 Free PMC article.
Cited by
-
Estrogen inhibits cardiomyocyte hypertrophy in vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP1.J Biol Chem. 2005 Jul 15;280(28):26339-48. doi: 10.1074/jbc.M414409200. Epub 2005 May 16. J Biol Chem. 2005. PMID: 15899894 Free PMC article.
-
The AP-1 transcription factor c-Jun prevents stress-imposed maladaptive remodeling of the heart.PLoS One. 2013 Sep 10;8(9):e73294. doi: 10.1371/journal.pone.0073294. eCollection 2013. PLoS One. 2013. PMID: 24039904 Free PMC article.
-
Reciprocal regulation of transcription factors and PLC isozyme gene expression in adult cardiomyocytes.J Cell Mol Med. 2010 Jun;14(6B):1824-35. doi: 10.1111/j.1582-4934.2009.00812.x. Epub 2009 Jun 16. J Cell Mol Med. 2010. PMID: 19538471 Free PMC article.
-
Direct interaction and reciprocal regulation between ASK1 and calcineurin-NFAT control cardiomyocyte death and growth.Mol Cell Biol. 2006 May;26(10):3785-97. doi: 10.1128/MCB.26.10.3785-3797.2006. Mol Cell Biol. 2006. PMID: 16648474 Free PMC article.
-
Extracellular signal-regulated kinase 1/2-mediated phosphorylation of p300 enhances myosin heavy chain I/beta gene expression via acetylation of nuclear factor of activated T cells c1.Nucleic Acids Res. 2011 Aug;39(14):5907-25. doi: 10.1093/nar/gkr162. Epub 2011 Apr 15. Nucleic Acids Res. 2011. PMID: 21498542 Free PMC article.
References
-
- Aramburu, J., M. B. Yaffe, C. Lopez-Rodriguez, L. C. Cantley, P. G. Hogan, and A. Rao. 1999. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285:2129-2133. - PubMed
-
- Beals, C. R., C. M. Sheridan, C. W. Turck, P. Gardner, and G. R. Crabtree. 1997. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 275:1930-1934. - PubMed
-
- Braz, J. C., O. F. Bueno, Q. Liang, B. J. Wilkins, Y. S. Dai, S. Parsons, J. Braunwart, B. J. Glascock, R. Klevitsky, T. F. Kimball, T. E. Hewett, and J. D. Molkentin. 2003. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J. Clin. Investig. 111:1475-1486. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous