

The CRM1 nuclear export receptor controls pathological cardiac gene expression
- PMID: 15572669
- PMCID: PMC533968
- DOI: 10.1128/MCB.24.24.10636-10649.2004
The CRM1 nuclear export receptor controls pathological cardiac gene expression
Abstract
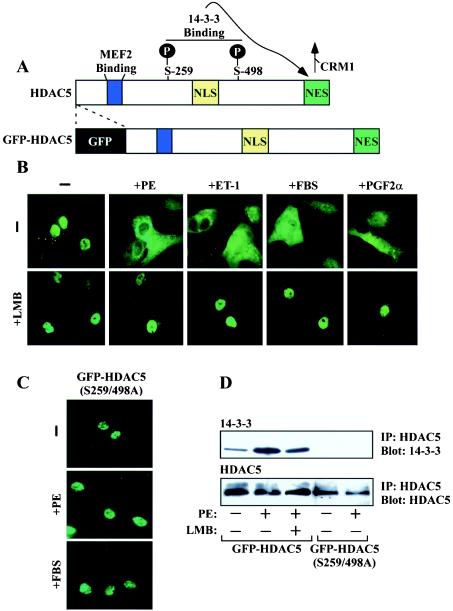
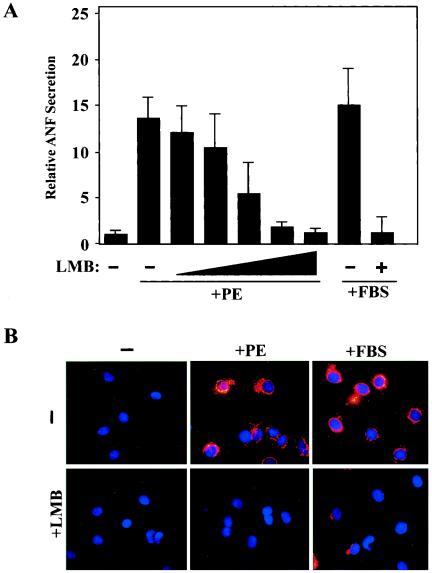
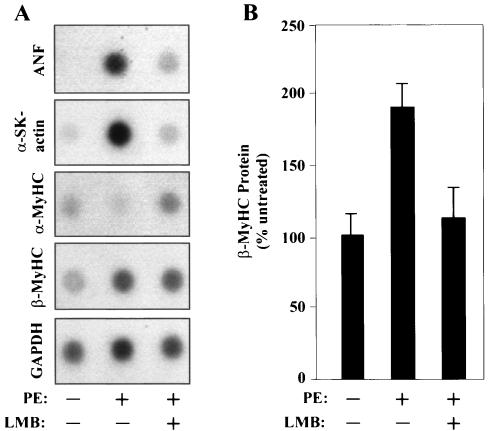
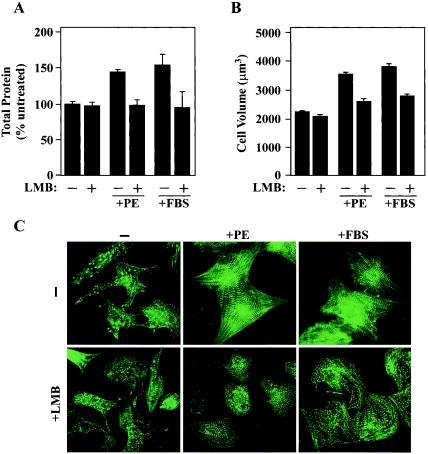
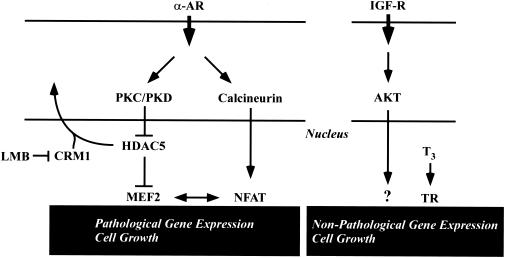
Diverse pathological insults trigger a cardiac remodeling process during which myocytes undergo hypertrophy, with consequent decline in cardiac function and eventual heart failure. Multiple transcriptional regulators of pathological cardiac hypertrophy are controlled at the level of subcellular distribution. For example, prohypertrophic transcription factors belonging to the nuclear factor of activated T cells (NFAT) and GATA families are subject to CRM1-dependent nuclear export but are rapidly relocalized to the nucleus in response to cues for hypertrophic growth. Here, we demonstrate that the antihypertrophic chromatin-modifying enzyme histone deacetylase 5 (HDAC5) is shuttled out of the cardiomyocyte nucleus via a CRM1-mediated pathway in response to diverse signals for hypertrophy. CRM1 antagonists block the agonist-mediated nuclear export of HDAC 5 and repress pathological gene expression and associated hypertrophy of cultured cardiomyocytes. Conversely, CRM1 activity is dispensable for nonpathological cardiac gene activation mediated by thyroid hormone and insulin-like growth factor 1, agonists that fail to trigger the nuclear export of HDAC5. These results suggest a selective role for CRM1 in derepression of pathological cardiac genes via its neutralizing effects on antihypertrophic factors such as HDAC5. Pharmacological approaches targeting CRM1-dependent nuclear export in heart muscle may have salutary effects on cardiac function by suppressing maladaptive changes in gene expression evoked by stress signals.
Figures
Similar articles
-
Suppression of HDAC nuclear export and cardiomyocyte hypertrophy by novel irreversible inhibitors of CRM1.Biochim Biophys Acta. 2009 May;1789(5):422-31. doi: 10.1016/j.bbagrm.2009.04.001. Epub 2009 May 3. Biochim Biophys Acta. 2009. PMID: 19414071
-
Neurohormonal regulation of cardiac histone deacetylase 5 nuclear localization by phosphorylation-dependent and phosphorylation-independent mechanisms.Circ Res. 2012 Jun 8;110(12):1585-95. doi: 10.1161/CIRCRESAHA.111.263665. Epub 2012 May 10. Circ Res. 2012. PMID: 22581927
-
Agonist-induced nuclear export of GFP-HDAC5 in isolated adult rat ventricular myocytes.J Pharmacol Toxicol Methods. 2009 May-Jun;59(3):135-40. doi: 10.1016/j.vascn.2009.03.002. Epub 2009 Mar 26. J Pharmacol Toxicol Methods. 2009. PMID: 19328241
-
Roles and post-translational regulation of cardiac class IIa histone deacetylase isoforms.J Physiol. 2015 Apr 15;593(8):1785-97. doi: 10.1113/jphysiol.2014.282442. Epub 2014 Nov 25. J Physiol. 2015. PMID: 25362149 Free PMC article. Review.
-
Nuclear Export as a Novel Therapeutic Target: The CRM1 Connection.Curr Cancer Drug Targets. 2015;15(7):575-92. doi: 10.2174/156800961507150828223554. Curr Cancer Drug Targets. 2015. PMID: 26324128 Review.
Cited by
-
Four-and-a-half LIM domains proteins are novel regulators of the protein kinase D pathway in cardiac myocytes.Biochem J. 2014 Feb 1;457(3):451-61. doi: 10.1042/BJ20131026. Biochem J. 2014. PMID: 24219103 Free PMC article.
-
Role of Mitochondrial RNA Polymerase in the Toxicity of Nucleotide Inhibitors of Hepatitis C Virus.Antimicrob Agents Chemother. 2015 Nov 23;60(2):806-17. doi: 10.1128/AAC.01922-15. Print 2016 Feb. Antimicrob Agents Chemother. 2015. PMID: 26596942 Free PMC article.
-
Toward transcriptional therapies for the failing heart: chemical screens to modulate genes.J Clin Invest. 2005 Mar;115(3):538-46. doi: 10.1172/JCI24144. J Clin Invest. 2005. PMID: 15765135 Free PMC article. Review.
-
HDAC5 promotes optic nerve regeneration by activating the mTOR pathway.Exp Neurol. 2019 Jul;317:271-283. doi: 10.1016/j.expneurol.2019.03.011. Epub 2019 Mar 22. Exp Neurol. 2019. PMID: 30910408 Free PMC article.
-
Alpha-adrenergic signalling activates protein kinase D and causes nuclear efflux of the transcriptional repressor HDAC5 in cultured adult mouse soleus skeletal muscle fibres.J Physiol. 2009 Mar 1;587(Pt 5):1101-15. doi: 10.1113/jphysiol.2008.164566. Epub 2009 Jan 5. J Physiol. 2009. PMID: 19124542 Free PMC article.
References
-
- Beals, C. R., N. A. Clipstone, S. N. Ho, and G. R. Crabtree. 1997. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 11:824-834. - PubMed
-
- Becker, T. C., R. J. Noel, W. S. Coats, A. M. Gomez-Foix, T. Alam, R. D. Gerard, and C. B. Newgard. 1994. Use of recombinant adenovirus for metabolic engineering of mammalian cells. Methods Cell Biol. 43:161-189. - PubMed
-
- Braunwald, E., and M. R. Bristow. 2000. Congestive heart failure: fifty years of progress. Circulation 102:IV14-IV23. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources