

Review
doi: 10.1038/sj.embor.7400250.
Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series
Affiliations
- PMID: 15459747
- PMCID: PMC1299150
- DOI: 10.1038/sj.embor.7400250
Item in Clipboard
Review
Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series
EMBO Rep.
2004 Oct.
Abstract
Huntington's disease (HD) is a late-onset neurodegenerative disorder that is caused by a CAG repeat expansion in the IT15 gene, which results in a long stretch of polyglutamine close to the amino-terminus of the HD protein huntingtin (htt). The normal function of htt, and the molecular mechanisms that contribute to the disease pathogenesis, are in the process of being elucidated. In this review, we outline the potential functions of htt as defined by the proteins with which it has been found to interact. We then focus on evidence that supports a role for transcriptional dysfunction and impaired protein folding and degradation as early events in disease pathogenesis.
Figures
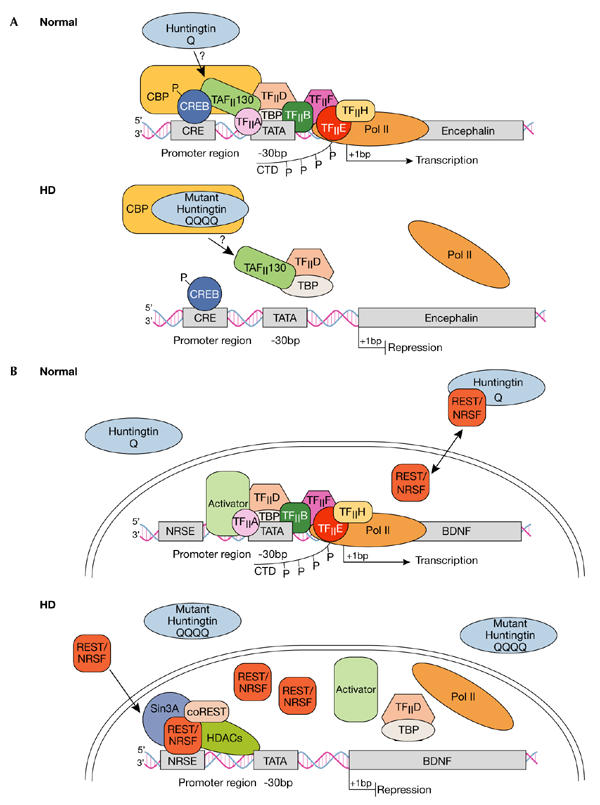
Dysregulation of cAMP-responsive-element- and NRSE-mediated transcription in Huntington's disease. (A) Normal: Huntingtin (htt) is predominantly located in the cytoplasm, although some reports indicate its presence in the nucleus. The transcription factor cAMP-responsive element (CRE)-binding protein (CREB) binds to DNA elements that contain CRE in cellular promoters such as in the encephalin gene, the activation of which is important in neuronal survival. Transcriptional activation is achieved by protein kinase A phosphorylation (P) of CREB, which allows for the subsequent recruitment of CREB-binding protein (CBP). CBP has intrinsic histone acetyltransferase activity, which remodels chromatin into an open architecture, allowing for the subsequent recruitment of the TAFII130 subunit of TFIID by CREB, and after this, the general transcriptional machinery, which includes transcription factors TFIIA, B, D, E, F and H, and TATA-binding protein (TBP). Once correctly targeted, RNA polymerase II (Pol II) is phosphorylated in its carboxy-terminal domain (CTD) by TFIIH to initiate transcription. HD: Mutant htt disrupts CRE-mediated transcription by directly interacting with or sequestering CBP, and possibly TAFII130, in aggregates in the nucleus. Ultimately, CBP and TAFII130 are prevented from binding to CRE regions in cellular promoters, so the general transcription apparatus along with Pol II are not correctly targeted to the promoter, so transcriptional activation is impaired. (B) Normal: The transcription factor REST–NRSF binds to DNA elements called NRSEs in neuronal gene promoters such as in the brain-derived neurotrophic factor (BDNF) gene. Wild-type htt sustains the production of BDNF, which is an important survival factor for the striatal neurons that die in HD, by interacting with REST–NRSF in the cytoplasm, thereby reducing its availability in the nucleus to bind to NRSE sites. Under these conditions, transcription of BDNF is promoted as activators can bind to the BDNF promoter elements and subsequently recruit the general transcriptional machinery and Pol II. HD: Mutant htt fails to interact with REST–NRSF in the cytoplasm, which leads to increased levels of REST–NRSF in the nucleus. Under these conditions, REST–NRSF binds avidly to the NRSE and promotes the recruitment of Sin3A–histone-deacetylase complexes (HDACs) that have histone deacetylase activity for remodelling chromatin into a closed architecture, thereby suppressing the transcription of BDNF. NRSE, neuron-restrictive silencer element; NRSF, neuron-restrictive silencer factor, REST, repressor-element-1 transcription factor.
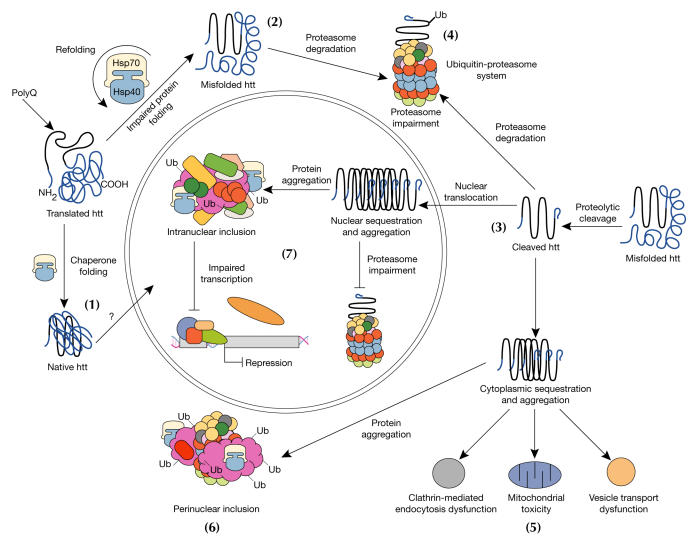
Model for cellular pathogenesis in Huntington's disease. The molecular chaperones Hsp70 and Hsp40 promote the folding of newly synthesized huntingtin (htt) into a native structure. Wild-type htt is predominantly cytoplasmic and probably functions in vesicle transport, cytoskeletal anchoring, clathrin-mediated endocytosis, neuronal transport or postsynaptic signalling. htt may be transported into the nucleus and have a role in transcriptional regulation (1). Chaperones can facilitate the recognition of abnormal proteins, promoting either their refolding, or ubiquitination (Ub) and subsequent degradation by the 26S proteasome. The HD mutation induces conformational changes and is likely to cause the abnormal folding of htt, which, if not corrected by chaperones, leads to the accumulation of misfolded htt in the cytoplasm (2). Alternatively, mutant htt might also be proteolytically cleaved, giving rise to amino-terminal fragments that form β-sheet structures (3). Ultimately, toxicity might be elicited by mutant full-length htt or by cleaved N-terminal fragments, which may form soluble monomers, oligomers or large insoluble aggregates. In the cytoplasm, mutant forms of htt may impair the ubiquitin–proteasome system (UPS), leading to the accumulation of more proteins that are misfolded (4). These toxic proteins might also impair normal vesicle transport and clathrin-mediated endocytosis. Also, the presence of mutant htt could activate proapoptotic proteins directly or indirectly by mitochondrial damage, leading to greater cellular toxicity and other deleterious effects (5). In an effort to protect itself, the cell accumulates toxic fragments into ubiquitinated cytoplasmic perinuclear aggregates (6). In addition, mutant htt can be translocated into the nucleus to form nuclear inclusions, which may disrupt transcription and the UPS (7).

Similar articles
-
Huntington's Disease.Cold Spring Harb Perspect Biol. 2011 Jun 1;3(6):a007476. doi: 10.1101/cshperspect.a007476. Cold Spring Harb Perspect Biol. 2011. PMID: 21441583 Free PMC article. Review.
-
Huntington's Disease: Mechanisms of Pathogenesis and Therapeutic Strategies.Cold Spring Harb Perspect Med. 2017 Jul 5;7(7):a024240. doi: 10.1101/cshperspect.a024240. Cold Spring Harb Perspect Med. 2017. PMID: 27940602 Free PMC article. Review.
-
PRMT5- mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington's disease (HD).Cell Cycle. 2015;14(11):1716-29. doi: 10.1080/15384101.2015.1033595. Cell Cycle. 2015. PMID: 25927346 Free PMC article.
-
Lessons from animal models of Huntington's disease.Trends Genet. 2002 Apr;18(4):202-9. doi: 10.1016/s0168-9525(01)02625-7. Trends Genet. 2002. PMID: 11932021 Review.
-
Huntington's disease: translating a CAG repeat into a pathogenic mechanism.Curr Opin Neurobiol. 1996 Oct;6(5):638-43. doi: 10.1016/s0959-4388(96)80097-3. Curr Opin Neurobiol. 1996. PMID: 8937828 Review.
Cited by
-
Caspase-6 does not contribute to the proteolysis of mutant huntingtin in the HdhQ150 knock-in mouse model of Huntington's disease.PLoS Curr. 2012 Jul 16;4:e4fd085bfc9973. doi: 10.1371/4fd085bfc9973. PLoS Curr. 2012. PMID: 22919566 Free PMC article. No abstract available.
-
Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease.J Biol Chem. 2010 Mar 19;285(12):8808-23. doi: 10.1074/jbc.M109.075028. Epub 2010 Jan 19. J Biol Chem. 2010. PMID: 20086007 Free PMC article.
-
Amphiphilic Nanocarrier Systems for Curcumin Delivery in Neurodegenerative Disorders.Medicines (Basel). 2018 Nov 23;5(4):126. doi: 10.3390/medicines5040126. Medicines (Basel). 2018. PMID: 30477087 Free PMC article. Review.
-
Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype.PLoS One. 2009 Sep 30;4(9):e7232. doi: 10.1371/journal.pone.0007232. PLoS One. 2009. PMID: 19789634 Free PMC article.
-
Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability.Biochim Biophys Acta Mol Basis Dis. 2019 Jun 1;1865(6):1428-1435. doi: 10.1016/j.bbadis.2019.02.015. Epub 2019 Feb 23. Biochim Biophys Acta Mol Basis Dis. 2019. PMID: 30802639 Free PMC article.
References
-
- Bates G (2003) Huntingtin aggregation and toxicity in Huntington's disease. Lancet 361: 1642–1644 - PubMed
-
- Bates GP, Murphy KP (2002) in Huntington's Disease (eds Bates GP, Harper PS, Jones AL) 387–426. Oxford, UK: Oxford University Press
-
- Bates GP, Harper PS, Jones AL (eds) (2002) Huntington's Disease. Oxford, UK: Oxford University Press.
-
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin–proteasome system by protein aggregation. Science 292: 1552–1555 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
