

Mutational dynamics of the SARS coronavirus in cell culture and human populations isolated in 2003
- PMID: 15347429
- PMCID: PMC517714
- DOI: 10.1186/1471-2334-4-32
Mutational dynamics of the SARS coronavirus in cell culture and human populations isolated in 2003
Abstract
Background: The SARS coronavirus is the etiologic agent for the epidemic of the Severe Acute Respiratory Syndrome. The recent emergence of this new pathogen, the careful tracing of its transmission patterns, and the ability to propagate in culture allows the exploration of the mutational dynamics of the SARS-CoV in human populations.
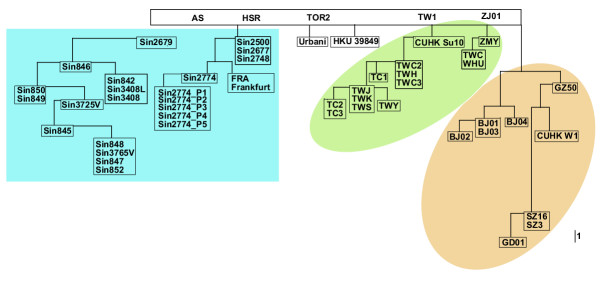
Methods: We sequenced complete SARS-CoV genomes taken from primary human tissues (SIN3408, SIN3725V, SIN3765V), cultured isolates (SIN848, SIN846, SIN842, SIN845, SIN847, SIN849, SIN850, SIN852, SIN3408L), and five consecutive Vero cell passages (SIN2774_P1, SIN2774_P2, SIN2774_P3, SIN2774_P4, SIN2774_P5) arising from SIN2774 isolate. These represented individual patient samples, serial in vitro passages in cell culture, and paired human and cell culture isolates. Employing a refined mutation filtering scheme and constant mutation rate model, the mutation rates were estimated and the possible date of emergence was calculated. Phylogenetic analysis was used to uncover molecular relationships between the isolates.
Results: Close examination of whole genome sequence of 54 SARS-CoV isolates identified before 14th October 2003, including 22 from patients in Singapore, revealed the mutations engendered during human-to-Vero and Vero-to-human transmission as well as in multiple Vero cell passages in order to refine our analysis of human-to-human transmission. Though co-infection by different quasipecies in individual tissue samples is observed, the in vitro mutation rate of the SARS-CoV in Vero cell passage is negligible. The in vivo mutation rate, however, is consistent with estimates of other RNA viruses at approximately 5.7 x 10-6 nucleotide substitutions per site per day (0.17 mutations per genome per day), or two mutations per human passage (adjusted R-square = 0.4014). Using the immediate Hotel M contact isolates as roots, we observed that the SARS epidemic has generated four major genetic groups that are geographically associated: two Singapore isolates, one Taiwan isolate, and one North China isolate which appears most closely related to the putative SARS-CoV isolated from a palm civet. Non-synonymous mutations are centered in non-essential ORFs especially in structural and antigenic genes such as the S and M proteins, but these mutations did not distinguish the geographical groupings. However, no non-synonymous mutations were found in the 3CLpro and the polymerase genes.
Conclusions: Our results show that the SARS-CoV is well adapted to growth in culture and did not appear to undergo specific selection in human populations. We further assessed that the putative origin of the SARS epidemic was in late October 2002 which is consistent with a recent estimate using cases from China. The greater sequence divergence in the structural and antigenic proteins and consistent deletions in the 3'--most portion of the viral genome suggest that certain selection pressures are interacting with the functional nature of these validated and putative ORFs.
Figures




Similar articles
-
Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection.Lancet. 2003 May 24;361(9371):1779-85. doi: 10.1016/s0140-6736(03)13414-9. Lancet. 2003. PMID: 12781537 Free PMC article.
-
Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination.J Virol. 2015 Oct;89(20):10532-47. doi: 10.1128/JVI.01048-15. Epub 2015 Aug 12. J Virol. 2015. PMID: 26269185 Free PMC article.
-
Isolation of virus from a SARS patient and genome-wide analysis of genetic mutations related to pathogenesis and epidemiology from 47 SARS-CoV isolates.Virus Genes. 2005 Jan;30(1):93-102. doi: 10.1007/s11262-004-4586-9. Virus Genes. 2005. PMID: 15744567 Free PMC article.
-
Molecular epidemiology, evolution and phylogeny of SARS coronavirus.Infect Genet Evol. 2019 Jul;71:21-30. doi: 10.1016/j.meegid.2019.03.001. Epub 2019 Mar 4. Infect Genet Evol. 2019. PMID: 30844511 Free PMC article. Review.
-
A review of studies on animal reservoirs of the SARS coronavirus.Virus Res. 2008 Apr;133(1):74-87. doi: 10.1016/j.virusres.2007.03.012. Epub 2007 Apr 23. Virus Res. 2008. PMID: 17451830 Free PMC article. Review.
Cited by
-
Isolation of ACE2-dependent and -independent sarbecoviruses from Chinese horseshoe bats.J Virol. 2023 Sep 28;97(9):e0039523. doi: 10.1128/jvi.00395-23. Epub 2023 Sep 1. J Virol. 2023. PMID: 37655938 Free PMC article.
-
An Overview on the Zoonotic Aspects of COVID-19.Proc Natl Acad Sci India Sect B Biol Sci. 2023 Apr 26:1-5. doi: 10.1007/s40011-023-01445-8. Online ahead of print. Proc Natl Acad Sci India Sect B Biol Sci. 2023. PMID: 37360152 Free PMC article. Review.
-
Phylogenetic landscape of Monkeypox Virus (MPV) during the early outbreak in New York City, 2022.Emerg Microbes Infect. 2023 Dec;12(1):e2192830. doi: 10.1080/22221751.2023.2192830. Emerg Microbes Infect. 2023. PMID: 36927408 Free PMC article.
-
Recycling of memory B cells between germinal center and lymph node subcapsular sinus supports affinity maturation to antigenic drift.Nat Commun. 2022 May 5;13(1):2460. doi: 10.1038/s41467-022-29978-y. Nat Commun. 2022. PMID: 35513371 Free PMC article.
-
SARS-CoV-2 host prediction based on virus-host genetic features.Sci Rep. 2022 Mar 17;12(1):4576. doi: 10.1038/s41598-022-08350-6. Sci Rep. 2022. PMID: 35301337 Free PMC article.
References
-
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ, SARS Working Group A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. New England Journal of Medicine. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. - DOI - PubMed
-
- Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. New England Journal of Medicine. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. - DOI - PubMed
-
- Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The Genome Sequence of the SARS-Associated Coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. - DOI - PubMed
-
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. - DOI - PubMed
-
- Ruan YJ, Wei CL, Ee AL, Vega VB, Thoreau H, Su ST, Chia JM, Ng P, Chiu KP, Lim L, Zhang T, Peng CK, Lin EO, Lee NM, Yee SL, Ng LF, Chee RE, Stanton LW, Long PM, Liu ET. Comparative Full-length Genome Sequence Analysis of 14 SARS Coronavirus Isolates and Common Mutations Associated with Putative Origins of Infection. Lancet. 2003;361:1779–1785. doi: 10.1016/S0140-6736(03)13414-9. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
