

Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity
- PMID: 15329404
- PMCID: PMC6729638
- DOI: 10.1523/JNEUROSCI.2002-04.2004
Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity
Abstract
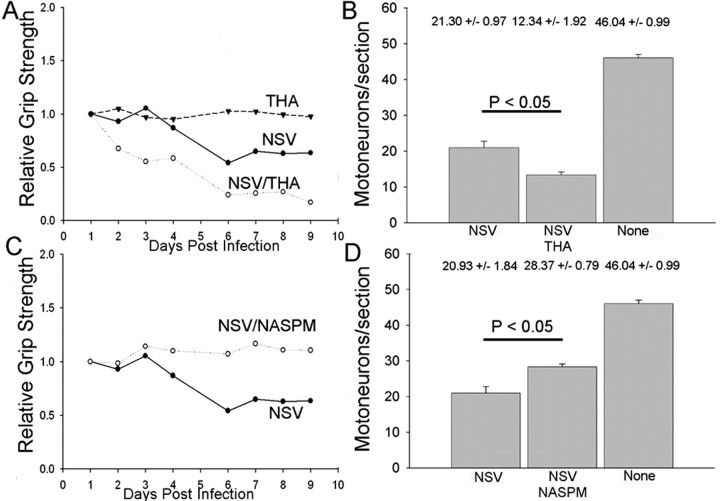
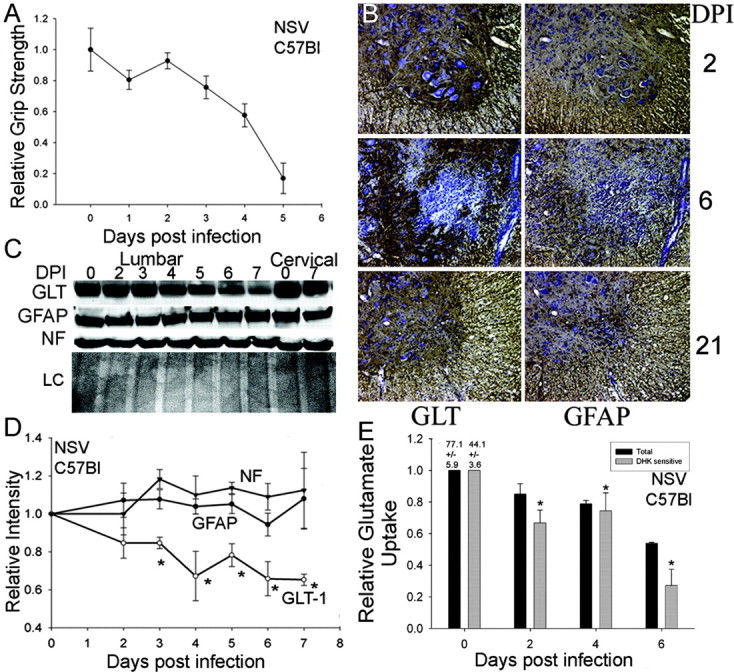
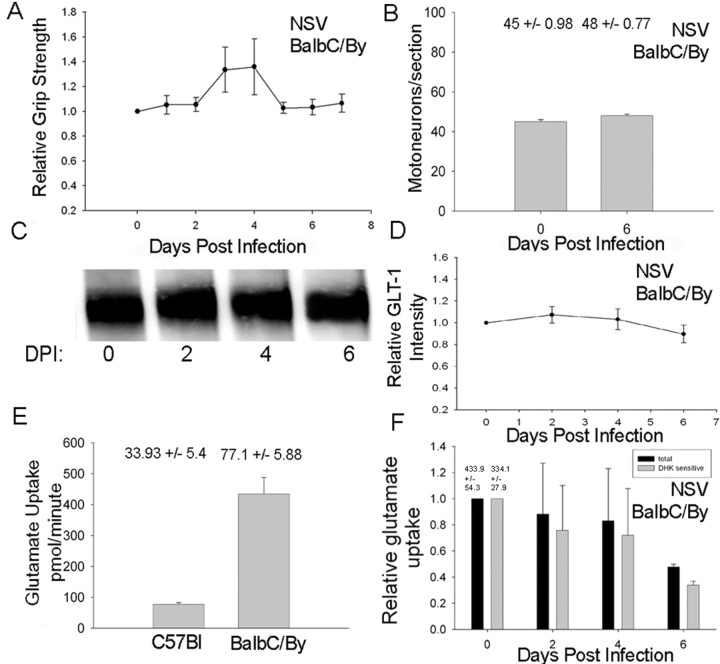
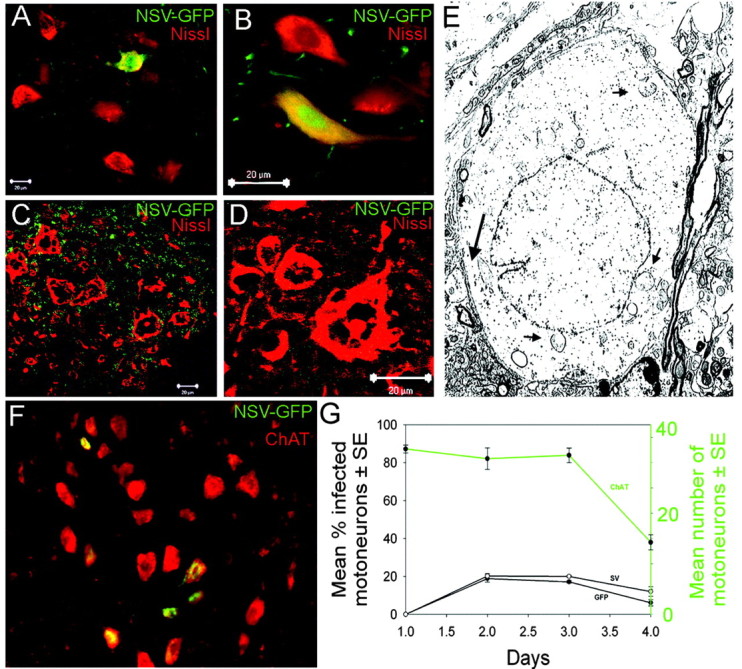
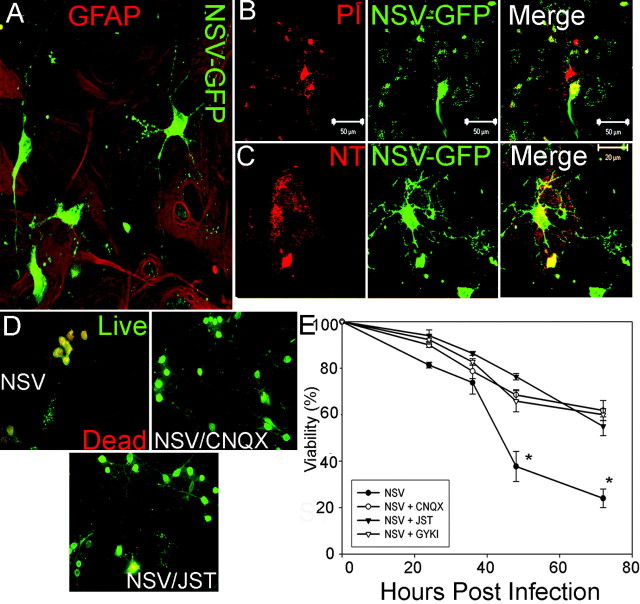
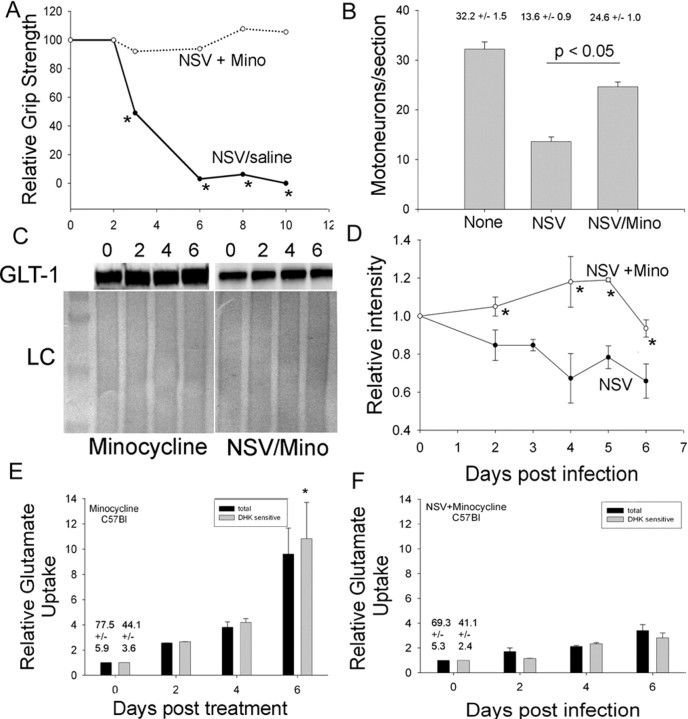
Neuroadapted Sindbis virus (NSV) is a neurotropic virus capable of inducing the death of spinal motor neurons in mice and rats. In this study we investigated the mechanisms that underlie NSV-induced motor neuron death. We found that many degenerating spinal motor neurons were not infected directly with NSV, suggesting that bystander cell death occurs. An excitotoxic mechanism was confirmed when blockade of calcium-permeable AMPA receptors attenuated motor neuron death both in vitro and in vivo. Blockade of astroglial glutamate reuptake potentiated NSV-induced motor neuron loss in vivo, suggesting that astrocyte-mediated removal of perisynaptic glutamate is important in limiting NSV-induced excitotoxic injury. Astroglial glutamate transport was reduced markedly in the spinal cord during NSV infection, in advance of motor neuron injury in susceptible mice. In contrast, we found 5.6-fold elevated glutamate uptake in the spinal cords of mice resistant to NSV-induced paralysis. Likewise, minocycline markedly increased spinal cord glutamate transport and protected mice from NSV-induced motor neuron death. These studies suggest that NSV infection triggers a cascade of events in the spinal cord resulting in impaired astrocytic glutamate transport and excitotoxic injury of motor neurons mediated via calcium-permeable AMPA receptors. Similar changes may occur in other motor neuron disorders such as amyotrophic lateral sclerosis or West Nile Virus-induced poliomyelitis, suggesting a common tissue injury pathway.
Figures
Similar articles
-
The opioid receptor antagonist, naloxone, protects spinal motor neurons in a murine model of alphavirus encephalomyelitis.Exp Neurol. 2007 Jun;205(2):461-70. doi: 10.1016/j.expneurol.2007.03.013. Epub 2007 Mar 23. Exp Neurol. 2007. PMID: 17459376 Free PMC article.
-
Tumor necrosis factor-alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis.Brain Res. 2009 Mar 31;1263:143-54. doi: 10.1016/j.brainres.2009.01.040. Epub 2009 Feb 3. Brain Res. 2009. PMID: 19368827 Free PMC article.
-
The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis.J Neurochem. 2008 May;105(4):1276-86. doi: 10.1111/j.1471-4159.2008.05230.x. Epub 2008 Jan 14. J Neurochem. 2008. PMID: 18194440 Free PMC article.
-
Alphavirus Encephalomyelitis: Mechanisms and Approaches to Prevention of Neuronal Damage.Neurotherapeutics. 2016 Jul;13(3):455-60. doi: 10.1007/s13311-016-0434-6. Neurotherapeutics. 2016. PMID: 27114366 Free PMC article. Review.
-
Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis.Muscle Nerve. 2002 Oct;26(4):438-58. doi: 10.1002/mus.10186. Muscle Nerve. 2002. PMID: 12362409 Review.
Cited by
-
The opioid receptor antagonist, naloxone, protects spinal motor neurons in a murine model of alphavirus encephalomyelitis.Exp Neurol. 2007 Jun;205(2):461-70. doi: 10.1016/j.expneurol.2007.03.013. Epub 2007 Mar 23. Exp Neurol. 2007. PMID: 17459376 Free PMC article.
-
Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases.mBio. 2014 Mar 11;5(2):e00902-14. doi: 10.1128/mBio.00902-14. mBio. 2014. PMID: 24618253 Free PMC article.
-
TNF-α triggers rapid membrane insertion of Ca(2+) permeable AMPA receptors into adult motor neurons and enhances their susceptibility to slow excitotoxic injury.Exp Neurol. 2012 Dec;238(2):93-102. doi: 10.1016/j.expneurol.2012.08.004. Epub 2012 Aug 19. Exp Neurol. 2012. PMID: 22921461 Free PMC article.
-
Rabies virus infection of primary neuronal cultures and adult mice: failure to demonstrate evidence of excitotoxicity.J Virol. 2006 Oct;80(20):10270-3. doi: 10.1128/JVI.01272-06. J Virol. 2006. PMID: 17005706 Free PMC article.
-
Delayed-onset and recurrent limb weakness associated with West Nile virus infection.J Neurovirol. 2010 Feb;16(1):93-100. doi: 10.3109/13550280903586378. J Neurovirol. 2010. PMID: 20166837
References
-
- Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32: 1-14. - PubMed
-
- Belin MF, Didier-Bazes M, Akaoka H, Hardin-Pouzet H, Bernard A, Giraudon P (1997) Changes in astrocytic glutamate catabolism enzymes following neuronal degeneration or viral infection. Glia 21: 154-161. - PubMed
-
- Biber K, Zuurman MW, Dijkstra IM, Boddeke HW (2002) Chemokines in the brain: neuroimmunology and beyond. Curr Opin Pharmacol 2: 63-68. - PubMed
-
- Binder GK, Griffin DE (2001) Interferon-γ-mediated site-specific clearance of alphavirus from CNS neurons. Science 293: 303-306. - PubMed
-
- Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR (1991) Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylate diastereomer. J Med Chem 34: 717-725. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources