

Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection
- PMID: 15263068
- PMCID: PMC503746
- DOI: 10.1073/pnas.0404310101
Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection
Abstract
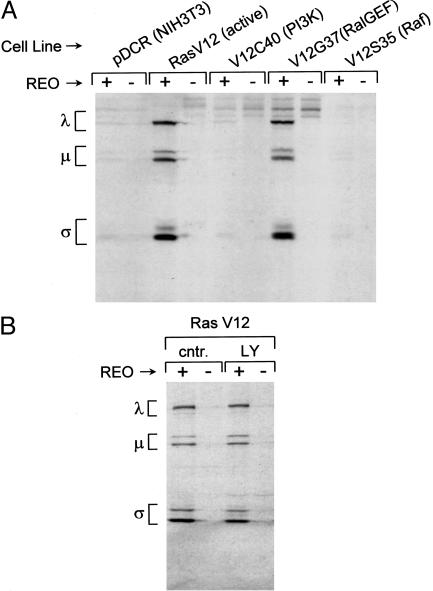
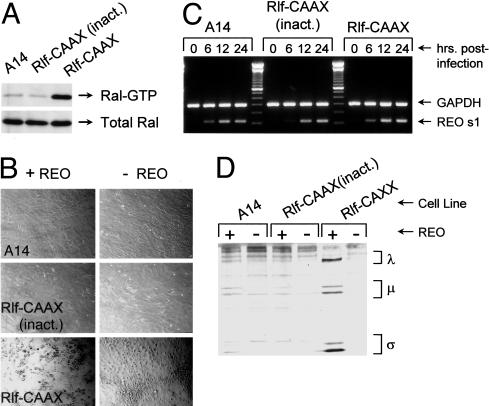
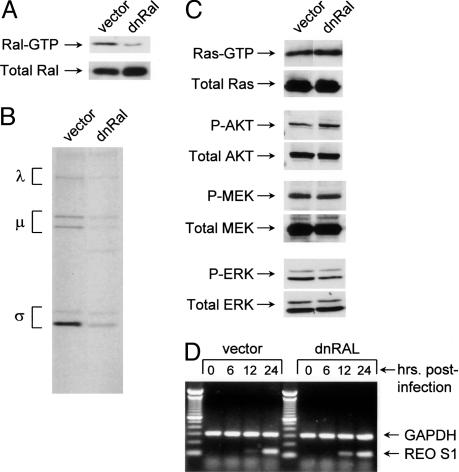
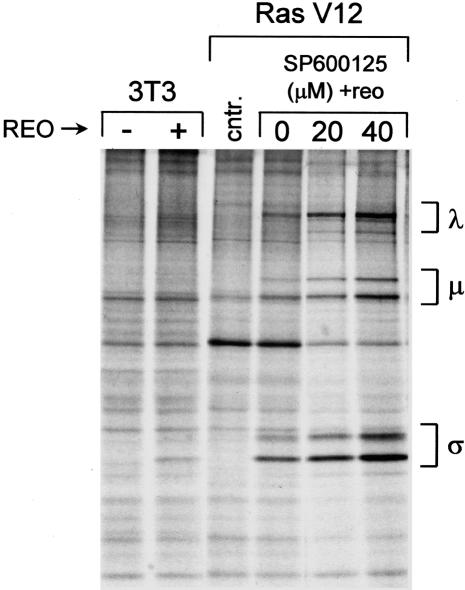
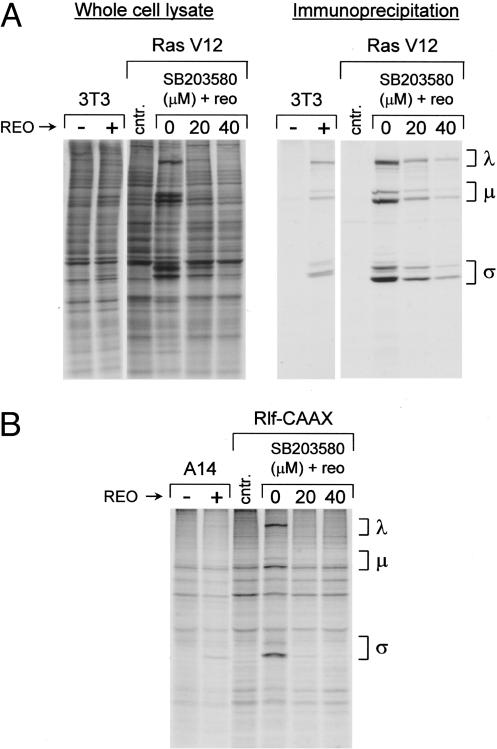
Reovirus is a benign human virus that was recently found to have oncolytic properties and is currently in clinical trials as a potential cancer therapy. We have previously demonstrated that activation of Ras signaling, a common event in cancer, renders cells susceptible to reovirus oncolysis. In this study, we investigate which elements downstream of Ras are important in reovirus infection. By using a panel of NIH 3T3 cells transformed with activated Ras mutated in the effector-binding domain, we found that only the RasV12G37 mutant, which was unable to signal to Raf or phosphatidylinositol 3-kinase but retained signaling capability to guanine nucleotide-exchange factors (GEFs) for the small G protein, Ral (known as RalGEFs), was permissive to reovirus. Expression of the activated mutant of the RalGEF, Rlf, also allowed reovirus replication. Specific inhibition of the Ral pathway by using dominant-negative RalA rendered normally permissive H-Ras cells (cells expressing activated Ras) resistant to reovirus. To further identify elements downstream of RalGEF that promote reovirus infection, we used chemical inhibitors of the downstream signaling elements p38 and JNK. We found that reovirus infection was blocked in the presence of the p38 inhibitor but not the JNK inhibitor. Together, these results implicate a Ras/RalGEF/p38 pathway in the regulation of reovirus replication and oncolysis.
Figures
Similar articles
-
Signal pathways which promote invasion and metastasis: critical and distinct contributions of extracellular signal-regulated kinase and Ral-specific guanine exchange factor pathways.Mol Cell Biol. 2001 Sep;21(17):5958-69. doi: 10.1128/MCB.21.17.5958-5969.2001. Mol Cell Biol. 2001. PMID: 11486034 Free PMC article.
-
Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation.Viral Immunol. 2005;18(1):89-115. doi: 10.1089/vim.2005.18.89. Viral Immunol. 2005. PMID: 15802955 Free PMC article. Review.
-
Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor-Ral pathway.Mol Cell Biol. 2000 Nov;20(22):8480-8. doi: 10.1128/MCB.20.22.8480-8488.2000. Mol Cell Biol. 2000. PMID: 11046144 Free PMC article.
-
Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer.Oncogene. 2005 Nov 21;24(52):7720-8. doi: 10.1038/sj.onc.1209041. Oncogene. 2005. PMID: 16299532 Review.
-
CUG2, a novel oncogene confers reoviral replication through Ras and p38 signaling pathway.Cancer Gene Ther. 2010 May;17(5):307-14. doi: 10.1038/cgt.2009.83. Epub 2010 Jan 15. Cancer Gene Ther. 2010. PMID: 20075984
Cited by
-
Activity levels of cathepsins B and L in tumor cells are a biomarker for efficacy of reovirus-mediated tumor cell killing.Cancer Gene Ther. 2015 Mar;22(4):188-97. doi: 10.1038/cgt.2015.4. Epub 2015 Jan 30. Cancer Gene Ther. 2015. PMID: 25633482
-
Anti-tumor efficacy of oncolytic reovirus against gastrointestinal stromal tumor cells.Oncotarget. 2017 Dec 18;8(70):115632-115646. doi: 10.18632/oncotarget.23361. eCollection 2017 Dec 29. Oncotarget. 2017. PMID: 29383187 Free PMC article.
-
Exploring Reovirus Plasticity for Improving Its Use as Oncolytic Virus.Viruses. 2015 Dec 24;8(1):4. doi: 10.3390/v8010004. Viruses. 2015. PMID: 26712782 Free PMC article. Review.
-
PUMA and NF-kB Are Cell Signaling Predictors of Reovirus Oncolysis of Breast Cancer.PLoS One. 2017 Jan 18;12(1):e0168233. doi: 10.1371/journal.pone.0168233. eCollection 2017. PLoS One. 2017. PMID: 28099441 Free PMC article.
-
Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer.Cell Death Dis. 2013 Jul 18;4(7):e728. doi: 10.1038/cddis.2013.259. Cell Death Dis. 2013. PMID: 23868061 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
