

The HIV protease inhibitor ritonavir blocks osteoclastogenesis and function by impairing RANKL-induced signaling
- PMID: 15254587
- PMCID: PMC449740
- DOI: 10.1172/JCI15797
The HIV protease inhibitor ritonavir blocks osteoclastogenesis and function by impairing RANKL-induced signaling
Abstract
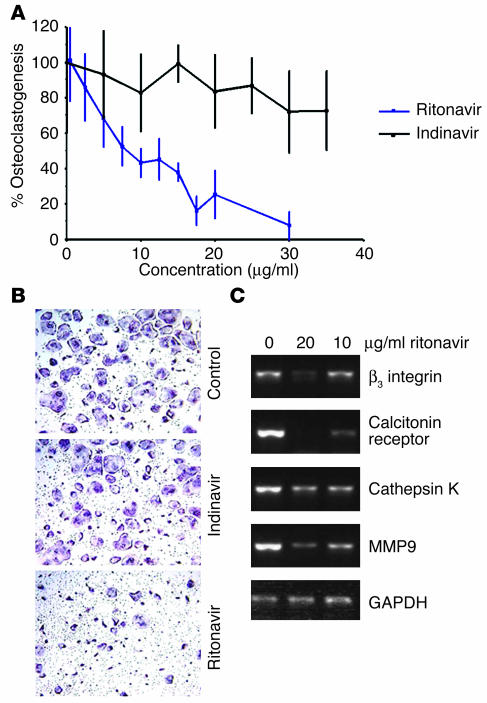
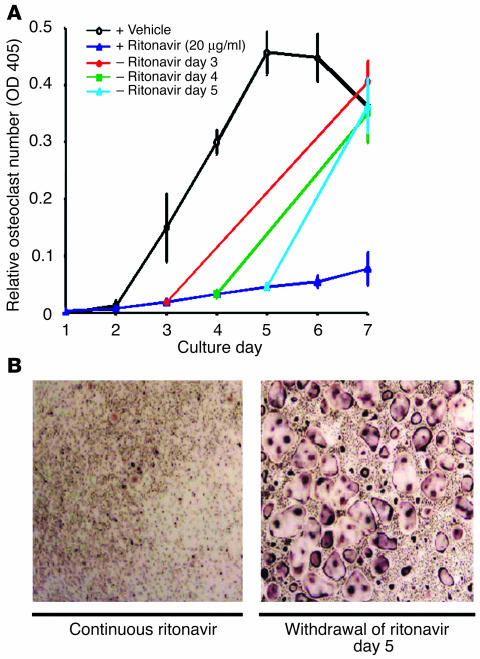
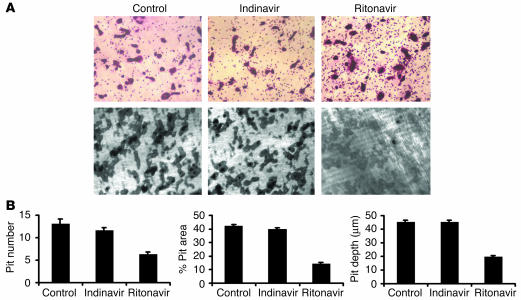
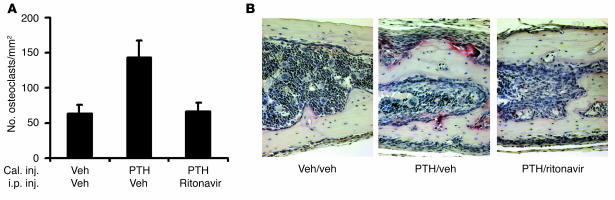
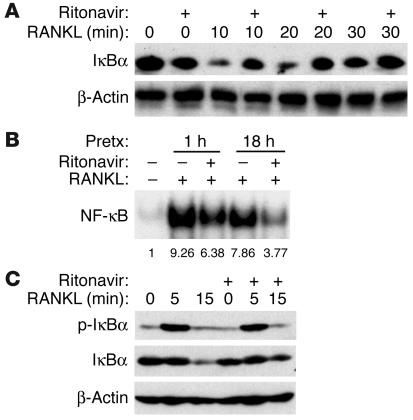
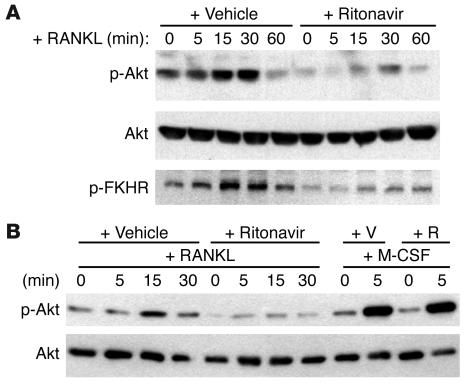
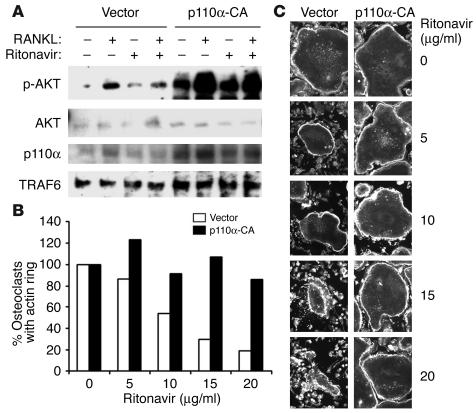
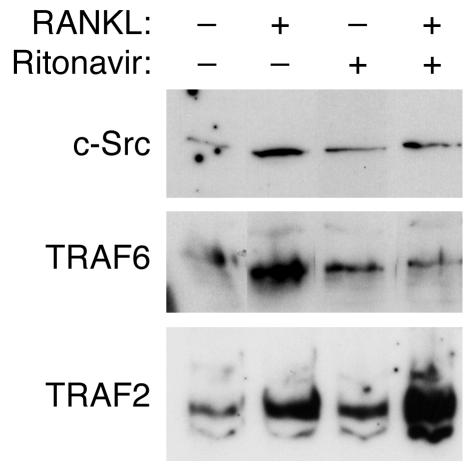
Highly active antiretroviral therapy (HAART), which includes HIV protease inhibitors (PIs), has been associated with bone demineralization. To determine if this complication reflects accelerated resorptive activity, we studied the impact of two common HIV PIs, ritonavir and indinavir, on osteoclast formation and function. Surprisingly, we find that ritonavir, but not indinavir, inhibits osteoclast differentiation in a reversible manner and also abrogates bone resorption by disrupting the osteoclast cytoskeleton, without affecting cell number. Ritonavir given in vivo completely blunts parathyroid hormone-induced osteoclastogenesis in mice, which confirms that the drug is bone sparing. In keeping with its antiresorptive properties, ritonavir impairs receptor activator of nuclear factor kappaB ligand-induced (RANKL-induced) activation of NF-kappaB and Akt signaling pathways, both critical to osteoclast formation and function. In particular, ritonavir is found to inhibit RANKL-induced Akt signaling by disrupting the recruitment of TNF receptor-associated factor 6/c-Src complex to lipid rafts. Thus, ritonavir may represent a bone-sparing PI capable of preventing development of osteopenia in patients currently on HAART.
Figures
Similar articles
-
Receptor activator of NF-kappa B ligand stimulates recruitment of SHP-1 to the complex containing TNFR-associated factor 6 that regulates osteoclastogenesis.J Immunol. 2003 Oct 1;171(7):3620-6. doi: 10.4049/jimmunol.171.7.3620. J Immunol. 2003. PMID: 14500659
-
Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors.J Cell Biochem. 2003 Sep 1;90(1):59-67. doi: 10.1002/jcb.10613. J Cell Biochem. 2003. PMID: 12938156
-
HIV envelope gp120-mediated regulation of osteoclastogenesis via receptor activator of nuclear factor kappa B ligand (RANKL) secretion and its modulation by certain HIV protease inhibitors through interferon-gamma/RANKL cross-talk.J Biol Chem. 2003 Nov 28;278(48):48251-8. doi: 10.1074/jbc.M304676200. Epub 2003 Sep 15. J Biol Chem. 2003. PMID: 12975380
-
Current topics in pharmacological research on bone metabolism: osteoclast differentiation regulated by glycosphingolipids.J Pharmacol Sci. 2006 Mar;100(3):195-200. doi: 10.1254/jphs.fmj05004x3. Epub 2006 Mar 14. J Pharmacol Sci. 2006. PMID: 16538029 Review.
-
Signal transduction pathways regulating osteoclast differentiation and function.J Bone Miner Metab. 2003;21(3):123-33. doi: 10.1007/s007740300021. J Bone Miner Metab. 2003. PMID: 12720046 Review. No abstract available.
Cited by
-
Influence of HIV Infection and Antiretroviral Therapy on Bone Homeostasis.Front Endocrinol (Lausanne). 2020 Sep 2;11:502. doi: 10.3389/fendo.2020.00502. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 32982960 Free PMC article. Review.
-
Bone Tissue Changes in Individuals Living with HIV/AIDS: The Importance of a Hierarchical Approach in Investigating Bone Fragility.J Pers Med. 2024 Jul 26;14(8):791. doi: 10.3390/jpm14080791. J Pers Med. 2024. PMID: 39201983 Free PMC article. Review.
-
The Role of Inflammatory Cytokines, the RANKL/OPG Axis, and the Immunoskeletal Interface in Physiological Bone Turnover and Osteoporosis.Scientifica (Cairo). 2013;2013:125705. doi: 10.1155/2013/125705. Epub 2013 Feb 3. Scientifica (Cairo). 2013. PMID: 24278766 Free PMC article. Review.
-
Role of T-cell reconstitution in HIV-1 antiretroviral therapy-induced bone loss.Nat Commun. 2015 Sep 22;6:8282. doi: 10.1038/ncomms9282. Nat Commun. 2015. PMID: 26392000 Free PMC article.
-
Src-like adaptor protein regulates osteoclast generation and survival.J Cell Biochem. 2010 May;110(1):201-9. doi: 10.1002/jcb.22527. J Cell Biochem. 2010. PMID: 20225239 Free PMC article.
References
-
- Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000;21:115–137. - PubMed
-
- Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. - PubMed
-
- Suda T, et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999;20:345–357. - PubMed
-
- Paton NIJ, Macallan DC, Griffin GE, Pazianas M. Bone mineral density in patients with human immunodeficiency virus infection. Calcif. Tissue Int. 1997;61:30–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- F32 AR008648/AR/NIAMS NIH HHS/United States
- AR-46852/AR/NIAMS NIH HHS/United States
- T32 AR007033/AR/NIAMS NIH HHS/United States
- R01 AR046523/AR/NIAMS NIH HHS/United States
- AR-48812/AR/NIAMS NIH HHS/United States
- R37 AR046523/AR/NIAMS NIH HHS/United States
- R01 AR046852/AR/NIAMS NIH HHS/United States
- U01 AI025903/AI/NIAID NIH HHS/United States
- AR-07033/AR/NIAMS NIH HHS/United States
- R01 AR032788/AR/NIAMS NIH HHS/United States
- R01 AR048812/AR/NIAMS NIH HHS/United States
- F32 AR-08648/AR/NIAMS NIH HHS/United States
- AR-32788/AR/NIAMS NIH HHS/United States
- AI-25903/AI/NIAID NIH HHS/United States
- AR-46523/AR/NIAMS NIH HHS/United States
- DE-05413/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
