

VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis
- PMID: 15229650
- PMCID: PMC514949
- DOI: 10.1038/sj.emboj.7600289
VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis
Abstract
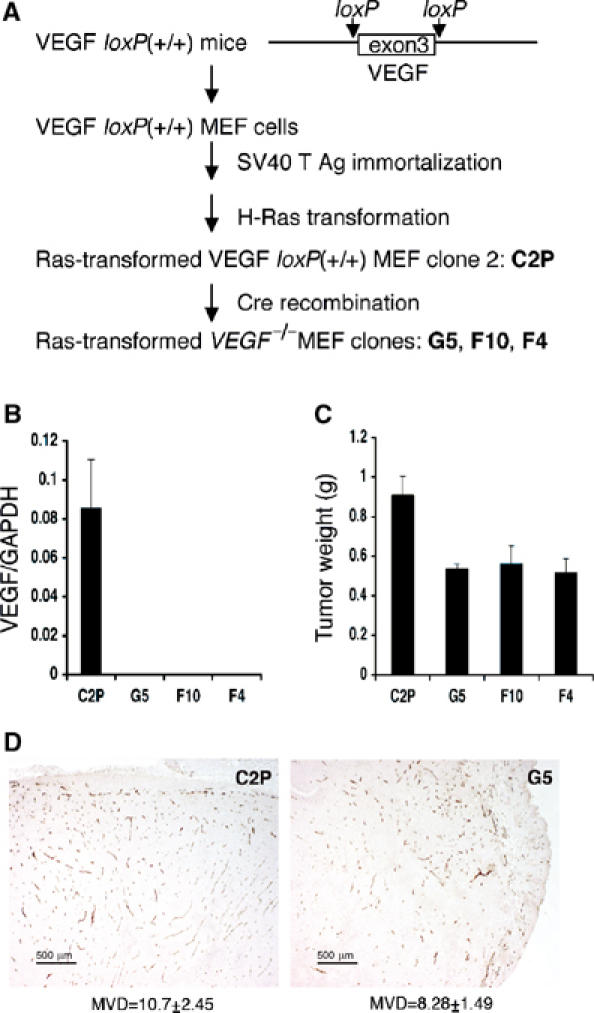
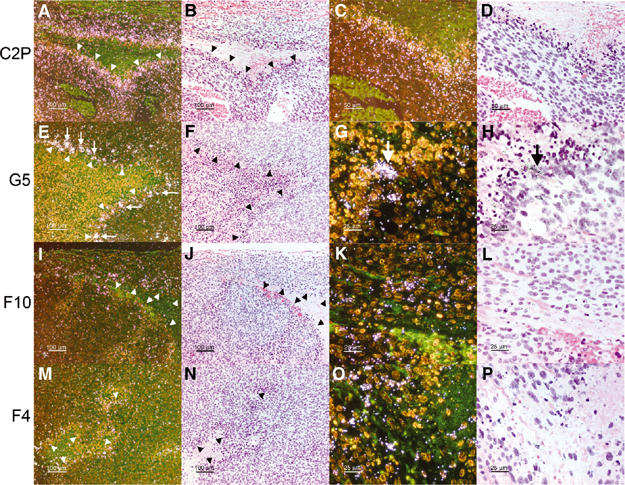
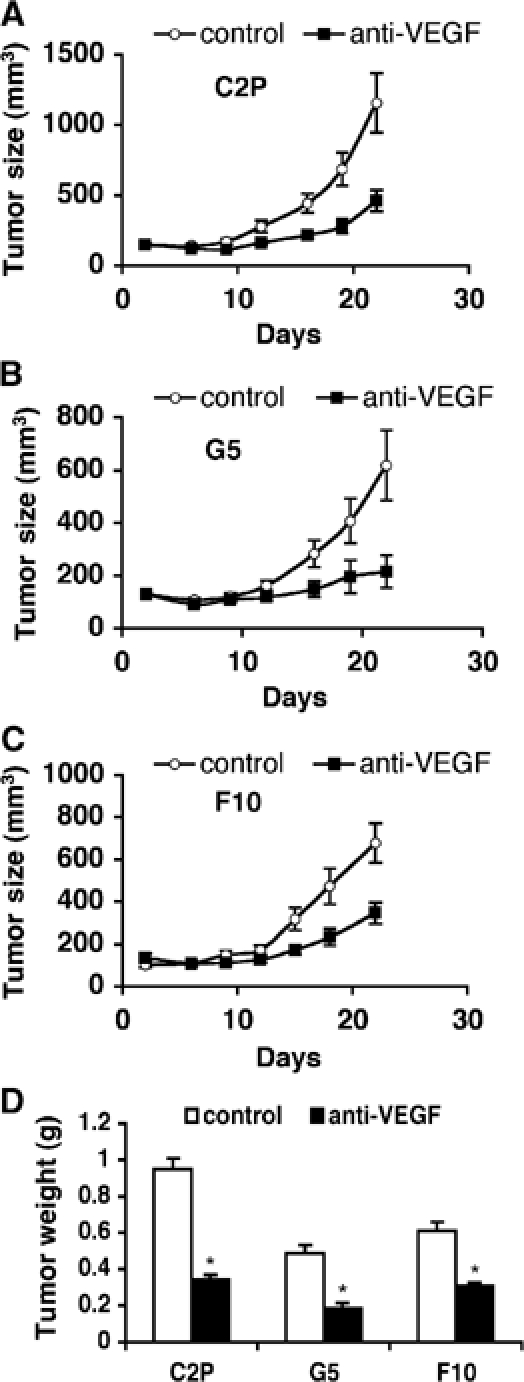
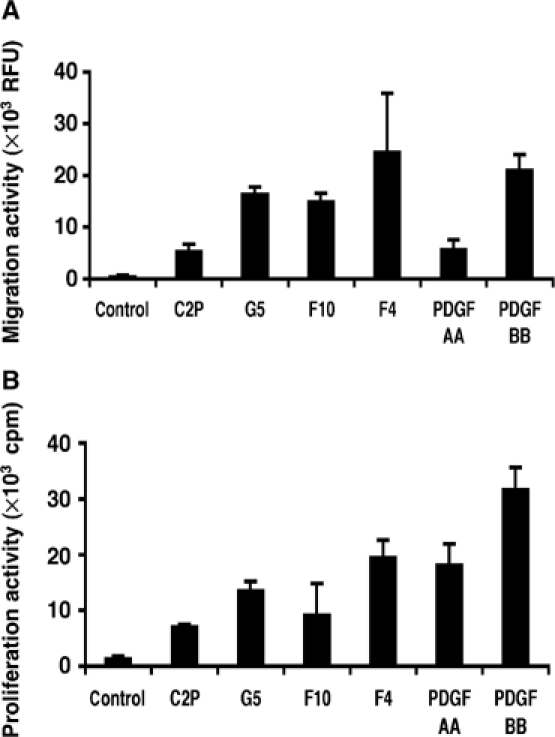
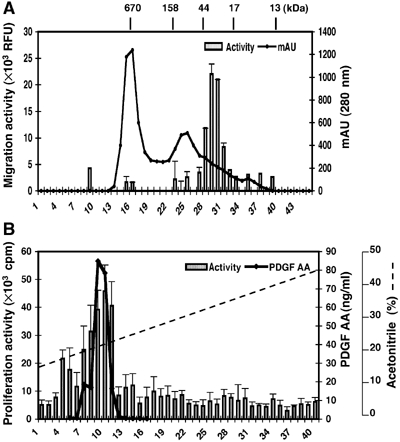
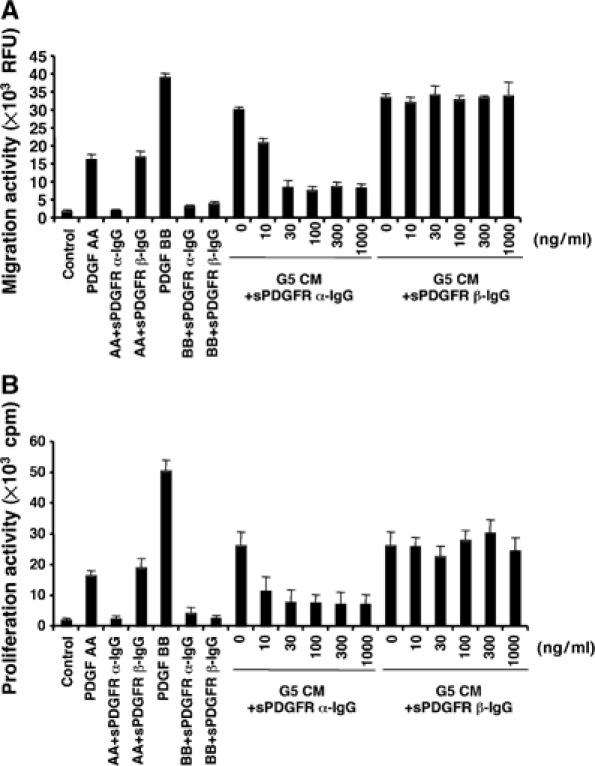
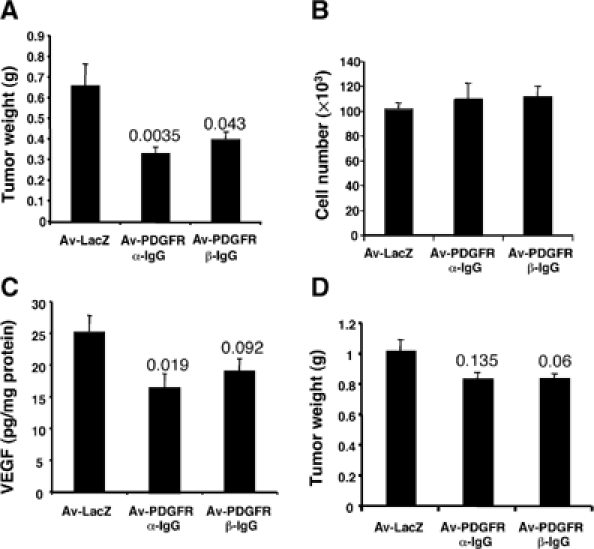
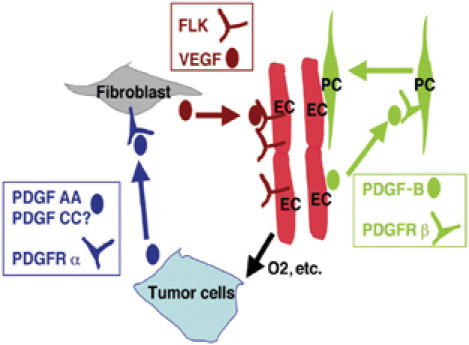
We generated VEGF-null fibrosarcomas from VEGF-loxP mouse embryonic fibroblasts to investigate the mechanisms of tumor escape after VEGF inactivation. These cells were found to be tumorigenic and angiogenic in vivo in spite of the absence of tumor-derived VEGF. However, VEGF derived from host stroma was readily detected in the tumor mass and treatment with a newly developed anti-VEGF monoclonal antibody substantially inhibited tumor growth. The functional significance of stroma-derived VEGF indicates that the recruitment of stromal cells is critical for the angiogenic and tumorigenic properties of these cells. Here we identified PDGF AA as the major stromal fibroblast chemotactic factor produced by tumor cells, and demonstrated that disrupting the paracrine PDGFR alpha signaling between tumor cells and stromal fibroblasts by soluble PDGFR alpha-IgG significantly reduced tumor growth. Thus, PDGFR alpha signaling is required for the recruitment of VEGF-producing stromal fibroblasts for tumor angiogenesis and growth. Our findings highlight a novel aspect of PDGFR alpha signaling in tumorigenesis.
Figures

Similar articles
-
Tumor-derived vascular endothelial growth factor up-regulates angiopoietin-2 in host endothelium and destabilizes host vasculature, supporting angiogenesis in ovarian cancer.Cancer Res. 2003 Jun 15;63(12):3403-12. Cancer Res. 2003. PMID: 12810677
-
PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment.Cancer Cell. 2009 Jan 6;15(1):21-34. doi: 10.1016/j.ccr.2008.12.004. Cancer Cell. 2009. PMID: 19111878
-
Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function.Cancer Res. 1999 Apr 1;59(7):1592-8. Cancer Res. 1999. PMID: 10197634
-
Tumor-stromal cell interactions and opportunities for therapeutic intervention.Curr Opin Pharmacol. 2010 Aug;10(4):369-74. doi: 10.1016/j.coph.2010.06.010. Epub 2010 Jul 16. Curr Opin Pharmacol. 2010. PMID: 20638903 Review.
-
Angiogenic and lymphangiogenic cascades in the tumor microenvironment.Front Biosci (Schol Ed). 2011 Jan 1;3(1):216-25. doi: 10.2741/s146. Front Biosci (Schol Ed). 2011. PMID: 21196371 Review.
Cited by
-
Serial measurements of serum PDGF-AA, PDGF-BB, FGF2, and VEGF in multiresistant ovarian cancer patients treated with bevacizumab.J Ovarian Res. 2012 Sep 19;5(1):23. doi: 10.1186/1757-2215-5-23. J Ovarian Res. 2012. PMID: 22989094 Free PMC article.
-
Choroid plexus papillomas: advances in molecular biology and understanding of tumorigenesis.Neuro Oncol. 2013 Mar;15(3):255-67. doi: 10.1093/neuonc/nos289. Epub 2012 Nov 21. Neuro Oncol. 2013. PMID: 23172371 Free PMC article. Review.
-
Growth factors in ischemic stroke.J Cell Mol Med. 2011 Aug;15(8):1645-87. doi: 10.1111/j.1582-4934.2009.00987.x. Epub 2009 Dec 8. J Cell Mol Med. 2011. PMID: 20015202 Free PMC article. Review.
-
Assessment of the platelet-derived growth factor receptor alpha antibody olaratumab in a panel of patient-derived soft tissue sarcoma xenografts.BMC Cancer. 2019 Jul 22;19(1):724. doi: 10.1186/s12885-019-5872-1. BMC Cancer. 2019. PMID: 31331295 Free PMC article.
-
Cancer-derived VEGF plays no role in malignant ascites formation in the mouse.World J Gastroenterol. 2005 Sep 21;11(35):5455-9. doi: 10.3748/wjg.v11.i35.5455. World J Gastroenterol. 2005. PMID: 16222736 Free PMC article.
References
-
- Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW (2002) Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res 62: 7042–7049 - PubMed
-
- Beckmann MP, Betsholtz C, Heldin CH, Westermark B, Di Marco E, Di Fiore PP, Robbins KC, Aaronson SA (1988) Comparison of biological properties and transforming potential of human PDGF-A and PDGF-B chains. Science 241: 1346–1349 - PubMed
-
- Berking C, Takemoto R, Schaider H, Showe L, Satyamoorthy K, Robbins P, Herlyn M (2001) Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res 61: 8306–8316 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
