

A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model
- PMID: 15146243
- PMCID: PMC406531
- DOI: 10.1172/JCI20864
A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model
Erratum in
- J Clin Invest. 2004 Aug;114(4):598
Abstract
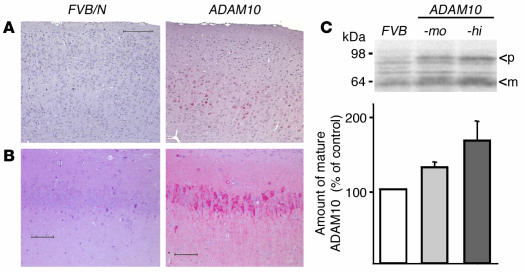
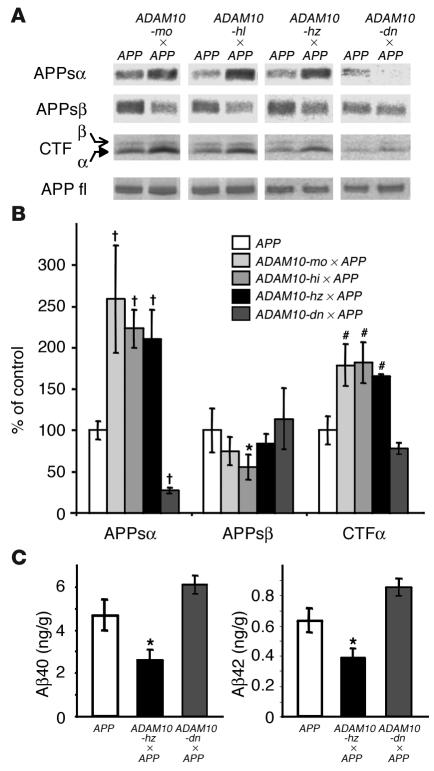
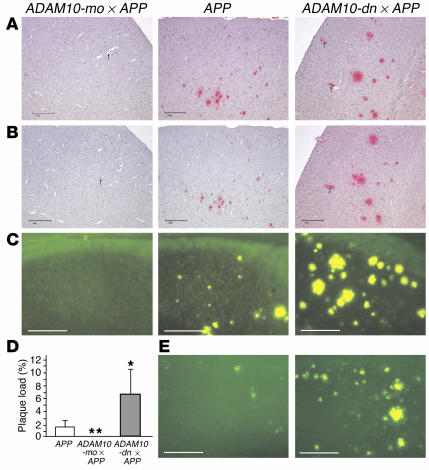
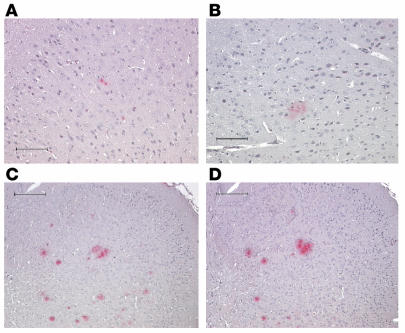
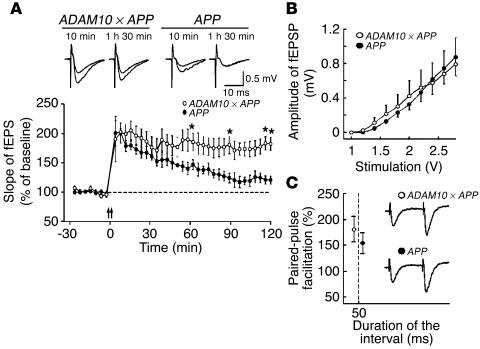
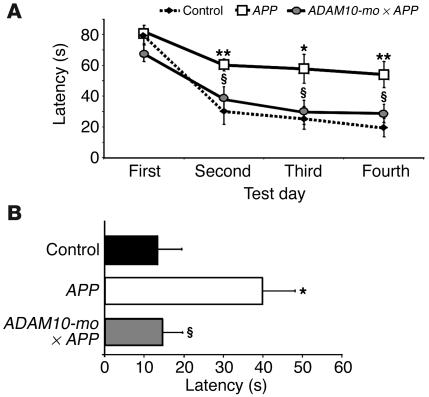
Alzheimer disease (AD) is characterized by excessive deposition of amyloid beta-peptides (A beta peptides) in the brain. In the nonamyloidogenic pathway, the amyloid precursor protein (APP) is cleaved by the alpha-secretase within the A beta peptide sequence. Proteinases of the ADAM family (adisintegrin and metalloproteinase) are the main candidates as physiologically relevant alpha-secretases, but early lethality of knockout animals prevented a detailed analysis in neuronal cells. To overcome this restriction, we have generated transgenic mice that overexpress either ADAM10 or a catalytically inactive ADAM10 mutant. In this report we show that a moderate neuronal overexpression of ADAM10 in mice transgenic for human APP([V717I]) increased the secretion of the neurotrophic soluble alpha-secretase-released N-terminal APP domain (APPs alpha), reduced the formation of A beta peptides, and prevented their deposition in plaques. Functionally, impaired long-term potentiation and cognitive deficits were alleviated. Expression of mutant catalytically inactive ADAM10 led to an enhancement of the number and size of amyloid plaques in the brains of double-transgenic mice. The results provide the first in vivo evidence for a proteinase of the ADAM family as an alpha-secretase of APP, reveal activation of ADAM10 as a promising therapeutic target, and support the hypothesis that a decrease in alpha-secretase activity contributes to the development of AD.
Figures
Comment in
-
Amyloid at the cutting edge: activation of alpha-secretase prevents amyloidogenesis in an Alzheimer disease mouse model.J Clin Invest. 2004 May;113(10):1384-7. doi: 10.1172/JCI21746. J Clin Invest. 2004. PMID: 15146234 Free PMC article. Review.
Similar articles
-
Effects of neuron-specific ADAM10 modulation in an in vivo model of acute excitotoxic stress.Neuroscience. 2008 Mar 18;152(2):459-68. doi: 10.1016/j.neuroscience.2007.10.060. Epub 2008 Jan 12. Neuroscience. 2008. PMID: 18276079
-
Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice.J Neurosci. 2002 May 1;22(9):3445-53. doi: 10.1523/JNEUROSCI.22-09-03445.2002. J Neurosci. 2002. PMID: 11978821 Free PMC article.
-
Amyloid at the cutting edge: activation of alpha-secretase prevents amyloidogenesis in an Alzheimer disease mouse model.J Clin Invest. 2004 May;113(10):1384-7. doi: 10.1172/JCI21746. J Clin Invest. 2004. PMID: 15146234 Free PMC article. Review.
-
Proteolytic processing of the Alzheimer's disease amyloid precursor protein in brain and platelets.J Neurosci Res. 2003 Nov 1;74(3):386-92. doi: 10.1002/jnr.10745. J Neurosci Res. 2003. PMID: 14598315
-
Alpha-secretase activation--an approach to Alzheimer's disease therapy.Neurodegener Dis. 2006;3(4-5):255-61. doi: 10.1159/000095264. Neurodegener Dis. 2006. PMID: 17047365 Review.
Cited by
-
An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin.Nat Neurosci. 2012 May;15(5):713-21. doi: 10.1038/nn.3080. Nat Neurosci. 2012. PMID: 22466506
-
Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer's Disease.Life (Basel). 2023 Nov 2;13(11):2156. doi: 10.3390/life13112156. Life (Basel). 2023. PMID: 38004296 Free PMC article.
-
GDE2-RECK controls ADAM10 α-secretase-mediated cleavage of amyloid precursor protein.Sci Transl Med. 2021 Mar 17;13(585):eabe6178. doi: 10.1126/scitranslmed.abe6178. Sci Transl Med. 2021. PMID: 33731436 Free PMC article.
-
The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model.Mol Psychiatry. 2017 Nov;22(11):1562-1575. doi: 10.1038/mp.2016.152. Epub 2016 Sep 20. Mol Psychiatry. 2017. PMID: 27646263 Free PMC article.
-
The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression.J Biol Chem. 2012 Nov 16;287(47):39753-65. doi: 10.1074/jbc.M112.416503. Epub 2012 Oct 3. J Biol Chem. 2012. PMID: 23035126 Free PMC article.
References
-
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. - PubMed
-
- Geula C, et al. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat. Med. 1998;4:827–831. - PubMed
-
- Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. - PubMed
-
- Gotz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ42 fibrils. Science. 2001;293:1491–1495. - PubMed
-
- Walsh DM, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
