

Alignment of protein sequences by their profiles
- PMID: 15044736
- PMCID: PMC2280052
- DOI: 10.1110/ps.03379804
Alignment of protein sequences by their profiles
Abstract
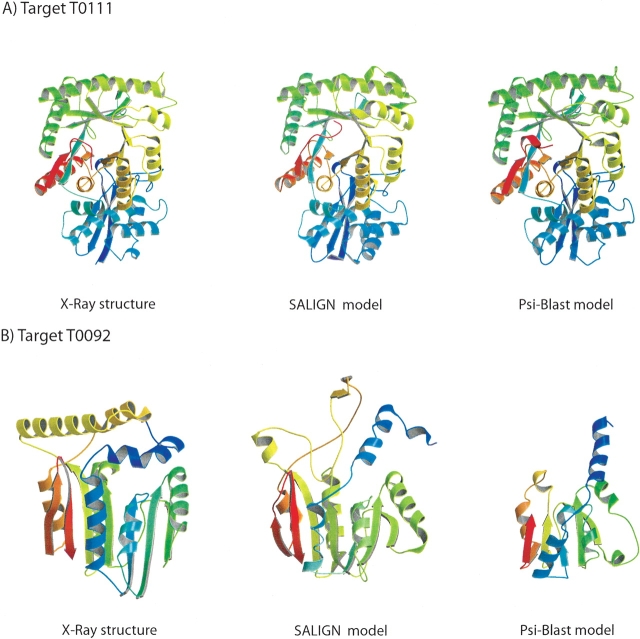
The accuracy of an alignment between two protein sequences can be improved by including other detectably related sequences in the comparison. We optimize and benchmark such an approach that relies on aligning two multiple sequence alignments, each one including one of the two protein sequences. Thirteen different protocols for creating and comparing profiles corresponding to the multiple sequence alignments are implemented in the SALIGN command of MODELLER. A test set of 200 pairwise, structure-based alignments with sequence identities below 40% is used to benchmark the 13 protocols as well as a number of previously described sequence alignment methods, including heuristic pairwise sequence alignment by BLAST, pairwise sequence alignment by global dynamic programming with an affine gap penalty function by the ALIGN command of MODELLER, sequence-profile alignment by PSI-BLAST, Hidden Markov Model methods implemented in SAM and LOBSTER, pairwise sequence alignment relying on predicted local structure by SEA, and multiple sequence alignment by CLUSTALW and COMPASS. The alignment accuracies of the best new protocols were significantly better than those of the other tested methods. For example, the fraction of the correctly aligned residues relative to the structure-based alignment by the best protocol is 56%, which can be compared with the accuracies of 26%, 42%, 43%, 48%, 50%, 49%, 43%, and 43% for the other methods, respectively. The new method is currently applied to large-scale comparative protein structure modeling of all known sequences.
Figures





Similar articles
-
Large-scale comparison of protein sequence alignment algorithms with structure alignments.Proteins. 2000 Jul 1;40(1):6-22. doi: 10.1002/(sici)1097-0134(20000701)40:1<6::aid-prot30>3.0.co;2-7. Proteins. 2000. PMID: 10813826
-
OXBench: a benchmark for evaluation of protein multiple sequence alignment accuracy.BMC Bioinformatics. 2003 Oct 10;4:47. doi: 10.1186/1471-2105-4-47. BMC Bioinformatics. 2003. PMID: 14552658 Free PMC article.
-
Simultaneous sequence alignment and tree construction using hidden Markov models.Pac Symp Biocomput. 2003:180-91. Pac Symp Biocomput. 2003. PMID: 12603027
-
Sequence comparison and protein structure prediction.Curr Opin Struct Biol. 2006 Jun;16(3):374-84. doi: 10.1016/j.sbi.2006.05.006. Epub 2006 May 19. Curr Opin Struct Biol. 2006. PMID: 16713709 Review.
-
Sequence and structure alignments in post-AlphaFold era.Curr Opin Struct Biol. 2023 Apr;79:102539. doi: 10.1016/j.sbi.2023.102539. Epub 2023 Feb 6. Curr Opin Struct Biol. 2023. PMID: 36753924 Review.
Cited by
-
An analysis of core deformations in protein superfamilies.Biophys J. 2005 Feb;88(2):1291-9. doi: 10.1529/biophysj.104.052449. Epub 2004 Nov 12. Biophys J. 2005. PMID: 15542556 Free PMC article.
-
An assessment of substitution scores for protein profile-profile comparison.Bioinformatics. 2011 Dec 15;27(24):3356-63. doi: 10.1093/bioinformatics/btr565. Epub 2011 Oct 13. Bioinformatics. 2011. PMID: 21998158 Free PMC article.
-
AlignHUSH: alignment of HMMs using structure and hydrophobicity information.BMC Bioinformatics. 2011 Jul 5;12:275. doi: 10.1186/1471-2105-12-275. BMC Bioinformatics. 2011. PMID: 21729312 Free PMC article.
-
Refinement by shifting secondary structure elements improves sequence alignments.Proteins. 2015 Mar;83(3):411-27. doi: 10.1002/prot.24746. Epub 2015 Jan 13. Proteins. 2015. PMID: 25546158 Free PMC article.
-
(PS)2-v2: template-based protein structure prediction server.BMC Bioinformatics. 2009 Oct 31;10:366. doi: 10.1186/1471-2105-10-366. BMC Bioinformatics. 2009. PMID: 19878598 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
