

JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease
- PMID: 14704277
- PMCID: PMC327205
- DOI: 10.1073/pnas.0307453101
JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease
Abstract
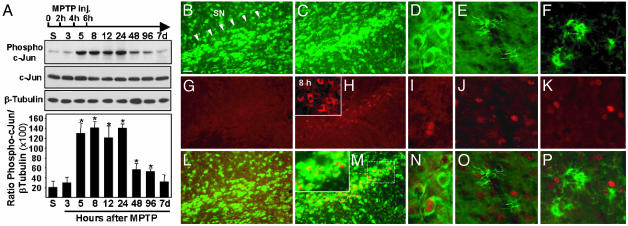
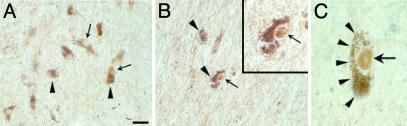
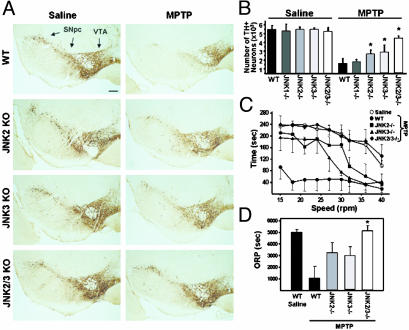
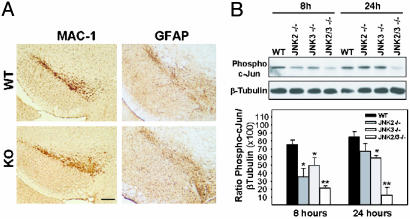
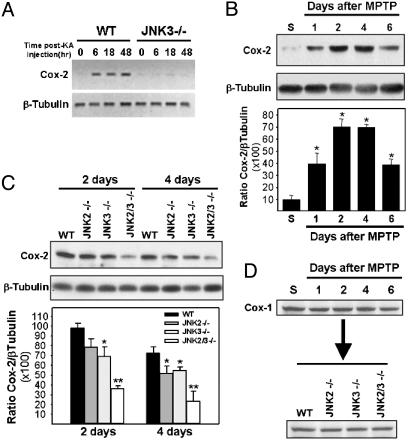
Parkinson's disease (PD) is a neurodegenerative disorder characterized by loss of dopamine-containing neurons, but the molecular pathways underlying its pathogenesis remain uncertain. Here, we show that by eliminating c-Jun N-terminal kinases (JNKs) we can prevent neurodegeneration and improve motor function in an animal model of PD. First, we found that c-Jun is activated in dopaminergic neurons from PD patients and in the 1-methyl-4-phenyl-1,2,4,6-tetrahydropyridine (MPTP) mouse model of PD. Examination of various JNK-deficient mice shows that both JNK2 and JNK3, but not JNK1, are required for MPTP-induced c-Jun activation and dopaminergic cell demise. Furthermore, we have identified cyclooxygenase (COX) 2 as a molecular target of JNK activation and demonstrated that COX-2 is indispensable for MPTP-induced dopaminergic cell death. Our data revealed that JNK2- and JNK3-induced COX-2 may be a principle pathway responsible for neurodegeneration in PD.
Figures
Similar articles
-
Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration.Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):5473-8. doi: 10.1073/pnas.0837397100. Epub 2003 Apr 17. Proc Natl Acad Sci U S A. 2003. PMID: 12702778 Free PMC article.
-
Cyclooxygenases mRNA and protein expression in striata in the experimental mouse model of Parkinson's disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration to mouse.Brain Res. 2004 Sep 3;1019(1-2):144-51. doi: 10.1016/j.brainres.2004.05.095. Brain Res. 2004. PMID: 15306248
-
Early signs of neuronal apoptosis in the substantia nigra pars compacta of the progressive neurodegenerative mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid model of Parkinson's disease.Neuroscience. 2006 Jun 19;140(1):67-76. doi: 10.1016/j.neuroscience.2006.02.007. Epub 2006 Mar 14. Neuroscience. 2006. PMID: 16533572
-
The role of c-Jun N-terminal kinase (JNK) in Parkinson's disease.IUBMB Life. 2003 Apr-May;55(4-5):267-71. doi: 10.1080/1521654031000121666. IUBMB Life. 2003. PMID: 12880208 Review.
-
From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance.IUBMB Life. 2005 Apr-May;57(4-5):283-95. doi: 10.1080/15216540500097111. IUBMB Life. 2005. PMID: 16036612 Review.
Cited by
-
Chronic Inflammation Links Cancer and Parkinson's Disease.Front Aging Neurosci. 2016 Jun 3;8:126. doi: 10.3389/fnagi.2016.00126. eCollection 2016. Front Aging Neurosci. 2016. PMID: 27375474 Free PMC article. Review.
-
Resveratrol potently reduces prostaglandin E2 production and free radical formation in lipopolysaccharide-activated primary rat microglia.J Neuroinflammation. 2007 Oct 10;4:25. doi: 10.1186/1742-2094-4-25. J Neuroinflammation. 2007. PMID: 17927823 Free PMC article.
-
Aminochrome as a preclinical experimental model to study degeneration of dopaminergic neurons in Parkinson's disease.Neurotox Res. 2007 Sep;12(2):125-34. doi: 10.1007/BF03033921. Neurotox Res. 2007. PMID: 17967736 Review.
-
JNK3 mediates paraquat- and rotenone-induced dopaminergic neuron death.J Neuropathol Exp Neurol. 2010 May;69(5):511-20. doi: 10.1097/NEN.0b013e3181db8100. J Neuropathol Exp Neurol. 2010. PMID: 20418776 Free PMC article.
-
Phosphoregulation on mitochondria: Integration of cell and organelle responses.CNS Neurosci Ther. 2019 Jul;25(7):837-858. doi: 10.1111/cns.13141. Epub 2019 Apr 25. CNS Neurosci Ther. 2019. PMID: 31025544 Free PMC article. Review.
References
-
- Fahn, S. & Przedborski, S. (2000). in Merritt's Neurology, ed. Rowland, L. P. (Lippincott Williams & Wilkins, New York), pp. 679–693.
-
- Lansbury, P. T., Jr., & Brice, A. (2002) Curr. Opin. Cell Biol. 14, 653–660. - PubMed
-
- Olanow, C. W. & Tatton, W. G. (1999) Annu. Rev. Neurosci. 22, 123–144. - PubMed
-
- Dauer, W. & Przedborski, S. (2003) Neuron 39, 889–909. - PubMed
-
- Beal, M. F. (1992) FASEB J. 6, 3338–3344. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
