

Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium
- PMID: 14681826
- PMCID: PMC1181897
- DOI: 10.1086/381000
Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium
Abstract
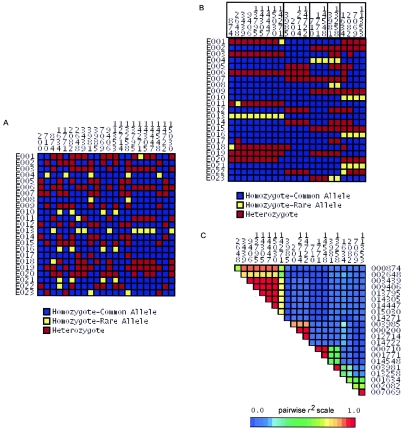
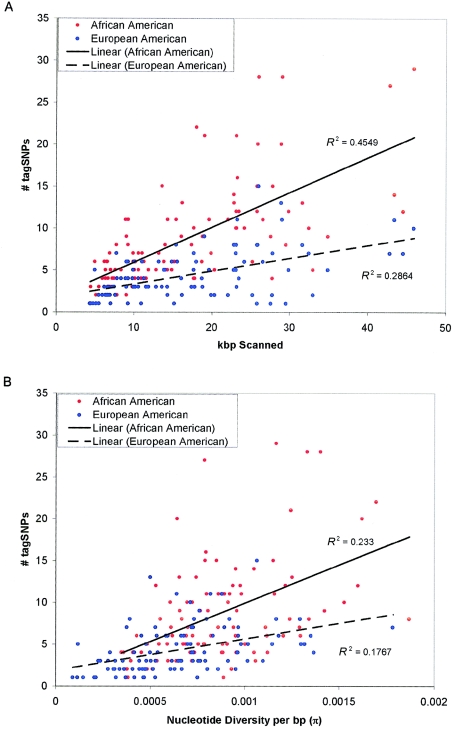
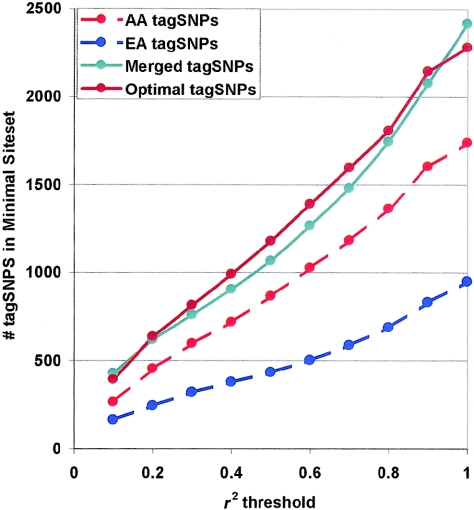
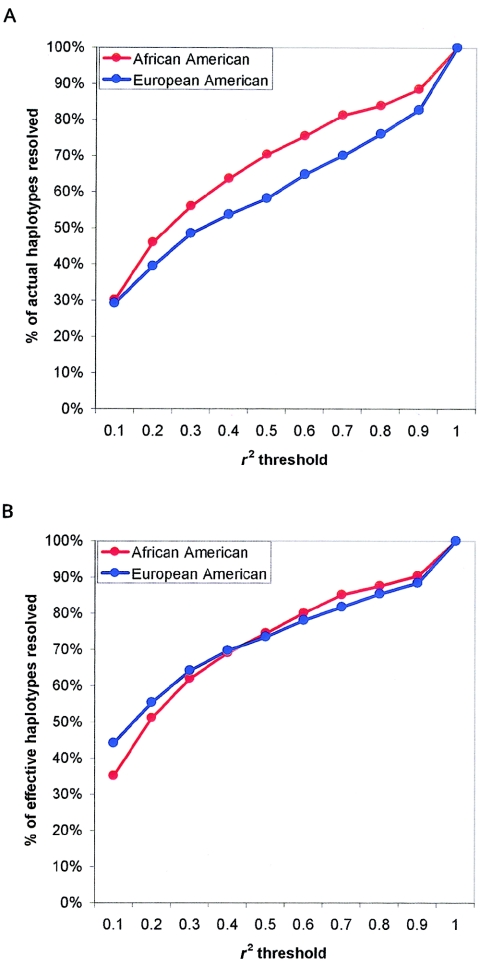
Common genetic polymorphisms may explain a portion of the heritable risk for common diseases. Within candidate genes, the number of common polymorphisms is finite, but direct assay of all existing common polymorphism is inefficient, because genotypes at many of these sites are strongly correlated. Thus, it is not necessary to assay all common variants if the patterns of allelic association between common variants can be described. We have developed an algorithm to select the maximally informative set of common single-nucleotide polymorphisms (tagSNPs) to assay in candidate-gene association studies, such that all known common polymorphisms either are directly assayed or exceed a threshold level of association with a tagSNP. The algorithm is based on the r(2) linkage disequilibrium (LD) statistic, because r(2) is directly related to statistical power to detect disease associations with unassayed sites. We show that, at a relatively stringent r(2) threshold (r2>0.8), the LD-selected tagSNPs resolve >80% of all haplotypes across a set of 100 candidate genes, regardless of recombination, and tag specific haplotypes and clades of related haplotypes in nonrecombinant regions. Thus, if the patterns of common variation are described for a candidate gene, analysis of the tagSNP set can comprehensively interrogate for main effects from common functional variation. We demonstrate that, although common variation tends to be shared between populations, tagSNPs should be selected separately for populations with different ancestries.
Figures
Similar articles
-
Efficient selection of tagging single-nucleotide polymorphisms in multiple populations.Hum Genet. 2006 Aug;120(1):58-68. doi: 10.1007/s00439-006-0182-5. Epub 2006 May 6. Hum Genet. 2006. PMID: 16680432
-
Similarity in recombination rate and linkage disequilibrium at CYP2C and CYP2D cytochrome P450 gene regions among Europeans indicates signs of selection and no advantage of using tagSNPs in population isolates.Pharmacogenet Genomics. 2012 Dec;22(12):846-57. doi: 10.1097/FPC.0b013e32835a3a6d. Pharmacogenet Genomics. 2012. PMID: 23089684
-
A comprehensive analysis of common genetic variation in prolactin (PRL) and PRL receptor (PRLR) genes in relation to plasma prolactin levels and breast cancer risk: the multiethnic cohort.BMC Med Genet. 2007 Dec 1;8:72. doi: 10.1186/1471-2350-8-72. BMC Med Genet. 2007. PMID: 18053149 Free PMC article.
-
On selecting markers for association studies: patterns of linkage disequilibrium between two and three diallelic loci.Genet Epidemiol. 2003 Jan;24(1):57-67. doi: 10.1002/gepi.10217. Genet Epidemiol. 2003. PMID: 12508256 Review.
-
[Linkage disequilibrium in the human genome and its exploitation].Arch Inst Pasteur Tunis. 2005;82(1-4):9-21. Arch Inst Pasteur Tunis. 2005. PMID: 16929750 Review. French.
Cited by
-
Common single nucleotide polymorphisms in genes related to immune function and risk of papillary thyroid cancer.PLoS One. 2013;8(3):e57243. doi: 10.1371/journal.pone.0057243. Epub 2013 Mar 8. PLoS One. 2013. PMID: 23520464 Free PMC article. Clinical Trial.
-
CUBN as a novel locus for end-stage renal disease: insights from renal transplantation.PLoS One. 2012;7(5):e36512. doi: 10.1371/journal.pone.0036512. Epub 2012 May 4. PLoS One. 2012. PMID: 22574174 Free PMC article.
-
GStream: improving SNP and CNV coverage on genome-wide association studies.PLoS One. 2013 Jul 3;8(7):e68822. doi: 10.1371/journal.pone.0068822. Print 2013. PLoS One. 2013. PMID: 23844243 Free PMC article.
-
Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy.Haematologica. 2015 Oct;100(10):1275-84. doi: 10.3324/haematol.2015.124875. Epub 2015 Jul 23. Haematologica. 2015. PMID: 26206798 Free PMC article.
-
Candidate gene studies in hypodontia suggest role for FGF3.Eur Arch Paediatr Dent. 2013 Dec;14(6):405-10. doi: 10.1007/s40368-013-0010-2. Epub 2013 Apr 3. Eur Arch Paediatr Dent. 2013. PMID: 23549991 Free PMC article.
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (Accession numbers for all genes are listed in .)
-
- HaploBlockFinder, http://cgi.uc.edu/cgi-bin/kzhang/haploBlockFinder.cgi/
-
- Pharmacogenetics and Risk of Cardiovascular Disease Project, http://droog.gs.washington.edu/parc/
-
- Phred/Phrap/Consed System Web Site, http://www.phrap.org/
References
-
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane CR, Lim EP, Kalayanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES (1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 22:231–23810.1038/10290 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
