

Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication
- PMID: 14585985
- PMCID: PMC262387
- DOI: 10.1128/MCB.23.22.8282-8294.2003
Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication
Abstract
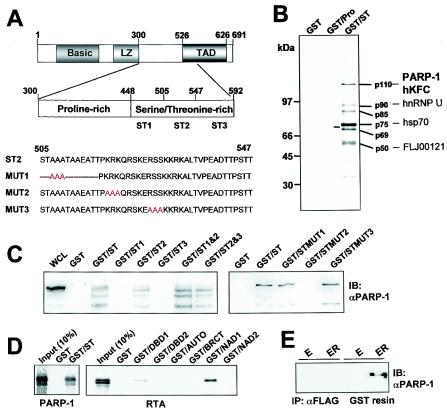
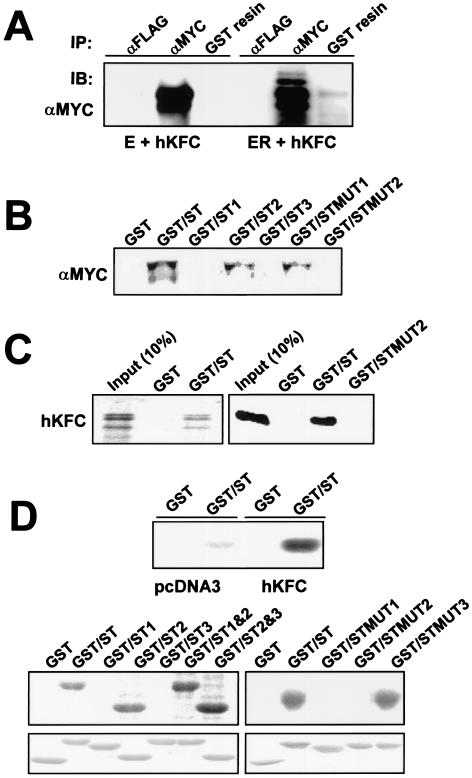
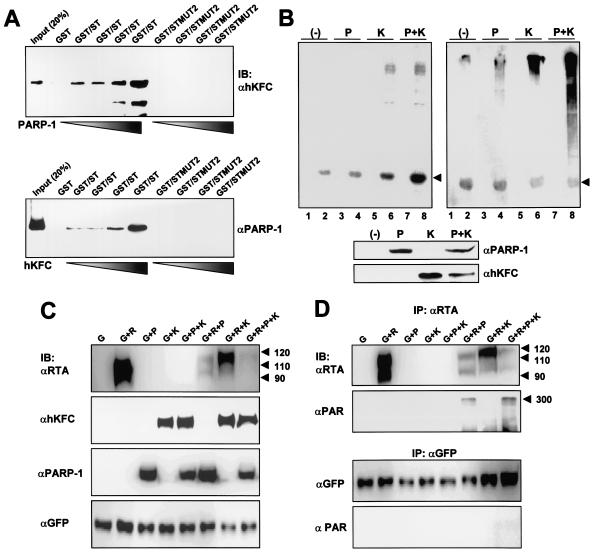
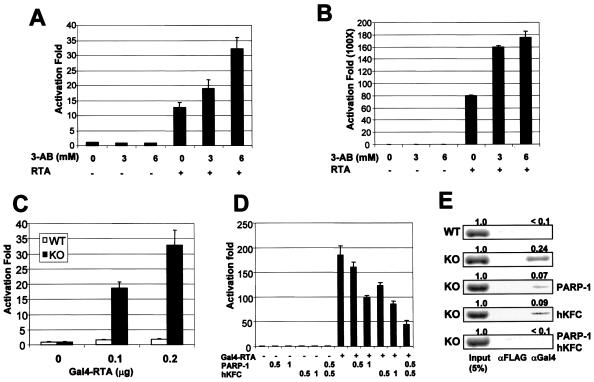
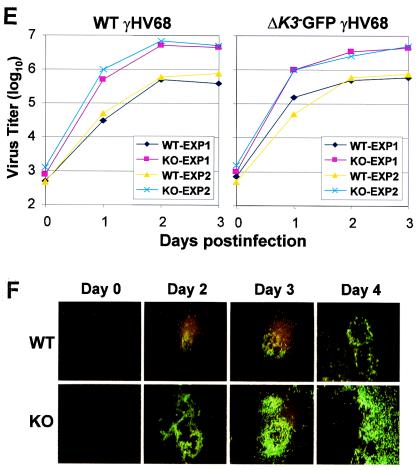
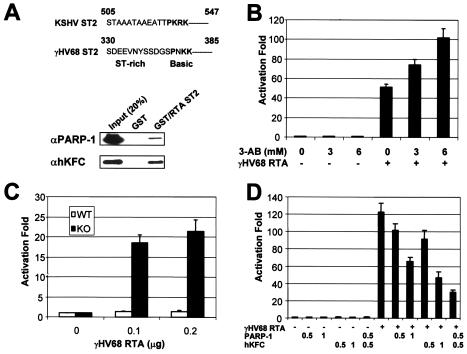
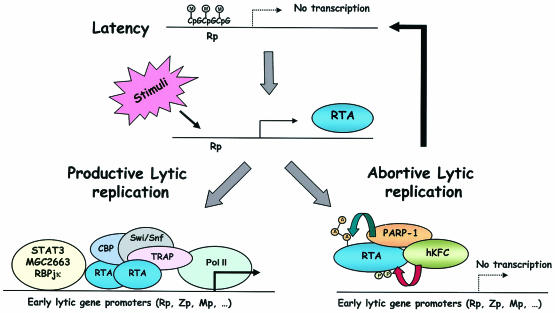
The replication and transcription activator (RTA) of gamma-2 herpesvirus is sufficient to drive the entire virus lytic cycle. Hence, the control of RTA activity should play an important role in the maintenance of viral latency. Here, we demonstrate that cellular poly(ADP-ribose) polymerase 1 (PARP-1) and Ste20-like kinase hKFC interact with the serine/threonine-rich region of gamma-2 herpesvirus RTA and that these interactions efficiently transfer poly(ADP-ribose) and phosphate units to RTA. Consequently, these modifications strongly repressed RTA-mediated transcriptional activation by inhibiting its recruitment onto the promoters of virus lytic genes. Conversely, the genetic ablation of PARP-1 and hKFC interaction or the knockout of the PARP-1 gene and activity considerably enhanced gamma-2 herpesvirus lytic replication. Thus, this is the first demonstration that cellular PARP-1 and hKFC act as molecular sensors to regulate RTA activity and thereby, herpesvirus latency.
Figures


Similar articles
-
Downregulation of Poly(ADP-Ribose) Polymerase 1 by a Viral Processivity Factor Facilitates Lytic Replication of Gammaherpesvirus.J Virol. 2015 Sep;89(18):9676-82. doi: 10.1128/JVI.00559-15. Epub 2015 Jul 8. J Virol. 2015. PMID: 26157130 Free PMC article.
-
Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency.J Virol. 2000 Apr;74(8):3659-67. doi: 10.1128/jvi.74.8.3659-3667.2000. J Virol. 2000. PMID: 10729142 Free PMC article.
-
Poly(ADP-ribose) polymerase 1 binds to Kaposi's sarcoma-associated herpesvirus (KSHV) terminal repeat sequence and modulates KSHV replication in latency.J Virol. 2004 Sep;78(18):9936-46. doi: 10.1128/JVI.78.18.9936-9946.2004. J Virol. 2004. PMID: 15331727 Free PMC article.
-
Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency.J Virol. 2004 Jun;78(12):6585-94. doi: 10.1128/JVI.78.12.6585-6594.2004. J Virol. 2004. PMID: 15163750 Free PMC article.
-
The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression.Oncogene. 2003 Aug 11;22(33):5150-63. doi: 10.1038/sj.onc.1206555. Oncogene. 2003. PMID: 12910252 Review.
Cited by
-
Ser-634 and Ser-636 of Kaposi's Sarcoma-Associated Herpesvirus RTA are Involved in Transactivation and are Potential Cdk9 Phosphorylation Sites.Front Microbiol. 2012 Feb 22;3:60. doi: 10.3389/fmicb.2012.00060. eCollection 2012. Front Microbiol. 2012. PMID: 22371709 Free PMC article.
-
KSHV Rta Promoter Specification and Viral Reactivation.Front Microbiol. 2012 Feb 14;3:30. doi: 10.3389/fmicb.2012.00030. eCollection 2012. Front Microbiol. 2012. PMID: 22347875 Free PMC article.
-
The virion-associated open reading frame 49 of murine gammaherpesvirus 68 promotes viral replication both in vitro and in vivo as a derepressor of RTA.J Virol. 2012 Jan;86(2):1109-18. doi: 10.1128/JVI.05785-11. Epub 2011 Nov 16. J Virol. 2012. PMID: 22090108 Free PMC article.
-
Interplay Between KSHV and the Host DNA Damage Response.Front Cell Infect Microbiol. 2020 Dec 9;10:604351. doi: 10.3389/fcimb.2020.604351. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 33425783 Free PMC article. Review.
-
ARID3B: a Novel Regulator of the Kaposi's Sarcoma-Associated Herpesvirus Lytic Cycle.J Virol. 2016 Sep 29;90(20):9543-55. doi: 10.1128/JVI.03262-15. Print 2016 Oct 15. J Virol. 2016. PMID: 27512077 Free PMC article.
References
-
- Bagrodia, S., and R. A. Cerione. 1999. Pak to the future. Trends Cell Biol. 9:350-355. - PubMed
-
- Bernstein, C., H. Bernstein, C. M. Payne, and H. Garewal. 2002. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat. Res. 511:145-178. - PubMed
-
- Boshoff, C., T. F. Schulz, M. M. Kennedy, A. K. Graham, C. Fisher, A. Thomas, J. O. McGee, R. A. Weiss, and J. J. O'Leary. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274-1278. - PubMed
-
- Buki, K. G., P. I. Bauer, A. Hakam, and E. Kun. 1995. Identification of domains of poly(ADP-ribose) polymerase for protein binding and self-association. J. Biol. Chem. 270:3370-3377. - PubMed
-
- Cervellera, M. N., and A. Sala. 2000. Poly(ADP-ribose) polymerase is a B-MYB coactivator. J. Biol. Chem. 275:10692-10696. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous