

IkappaB kinase-independent IkappaBalpha degradation pathway: functional NF-kappaB activity and implications for cancer therapy
- PMID: 14585967
- PMCID: PMC262380
- DOI: 10.1128/MCB.23.22.8070-8083.2003
IkappaB kinase-independent IkappaBalpha degradation pathway: functional NF-kappaB activity and implications for cancer therapy
Abstract
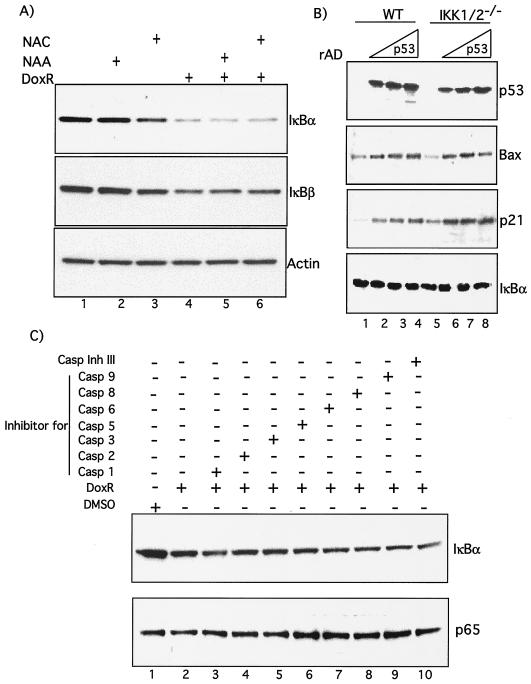
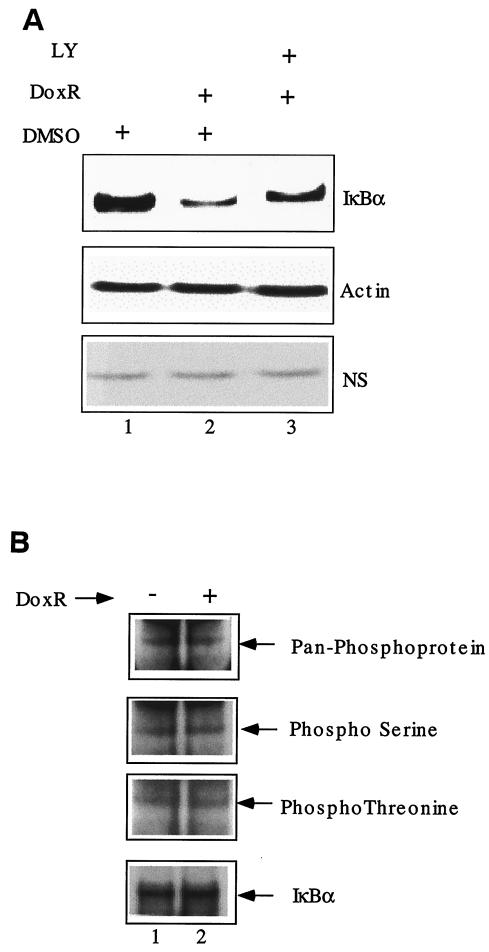
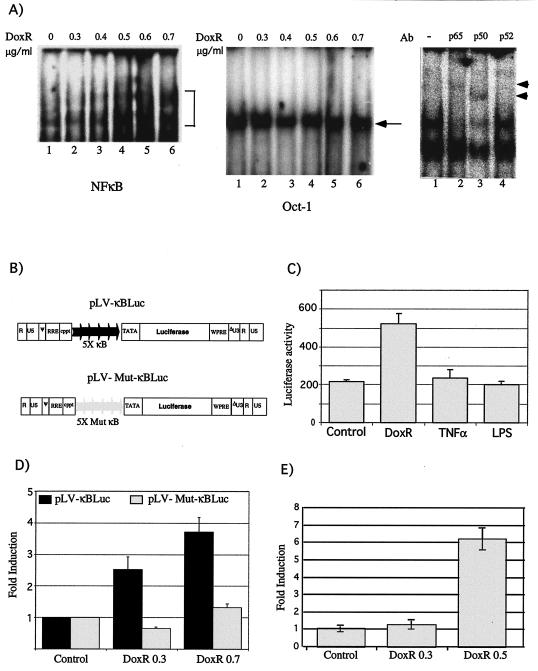
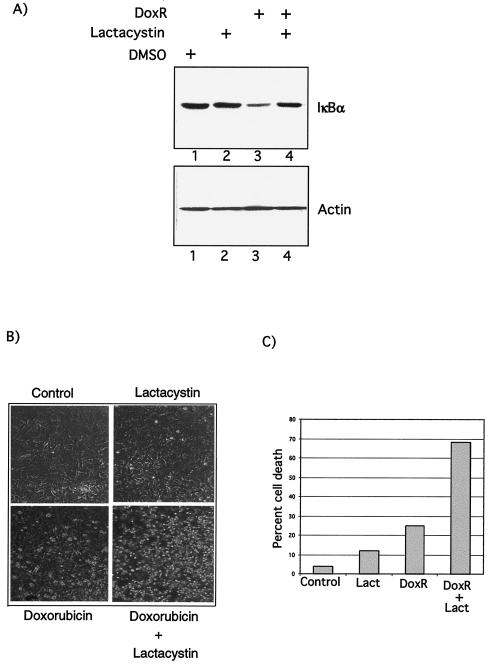
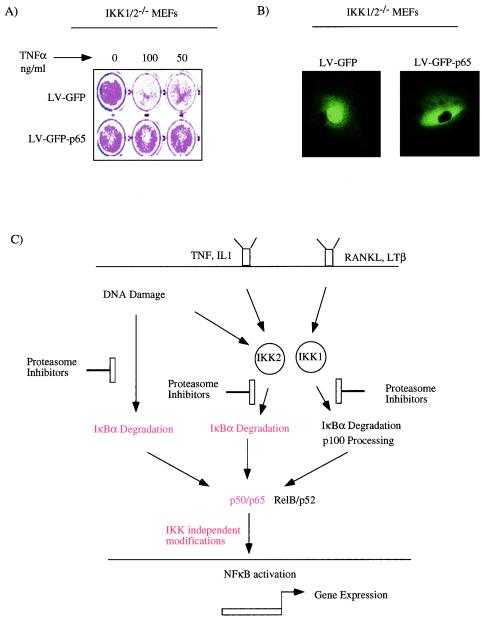
Antiapoptotic activity of NF-kappaB in tumors contributes to acquisition of resistance to chemotherapy. Degradation of IkappaB is a seminal step in activation of NF-kappaB. The IkappaB kinases, IKK1 and IKK2, have been implicated in both IkappaB degradation and subsequent modifications of NFkappaB. Using mouse embryo fibroblasts (MEFs) devoid of both IKK1 and IKK2 genes (IKK1/2(-/-)), we document a novel IkappaB degradation mechanism. We show that this degradation induced by a chemotherapeutic agent, doxorubicin (DoxR), does not require the classical serine 32 and 36 phosphorylation or the PEST domain of IkappaBalpha. Degradation of IkappaBalpha is partially blocked by phosphatidylinositol 3-kinase inhibitor LY294002 and is mediated by the proteasome. Free NF-kappaB generated by DoxR-induced IkappaB degradation in IKK1/2(-/-) cells is able to activate chromatin based NF-kappaB reporter gene and expression of the endogenous target gene, IkappaBalpha. These results also imply that modification of NF-kappaB by IKK1 or IKK2 either prior or subsequent to its release from IkappaB is not essential for NF-kappaB-mediated gene expression at least in response to DNA damage. In addition, DoxR-induced cell death in IKK1/2(-/-) MEFs is enhanced by simultaneous inhibition of NF-kappaB activation by blocking the proteasome activity. These results reveal an additional pathway of activating NF-kappaB during the course of anticancer therapy and provide a mechanistic basis for the observation that proteasome inhibitors could be used as adjuvants in chemotherapy.
Figures
Similar articles
-
Human T-cell lymphotropic virus type 1 tax induction of biologically Active NF-kappaB requires IkappaB kinase-1-mediated phosphorylation of RelA/p65.J Biol Chem. 2004 Apr 30;279(18):18137-45. doi: 10.1074/jbc.M401397200. Epub 2004 Feb 12. J Biol Chem. 2004. PMID: 14963024
-
Proteasome inhibitors induce inhibitory kappa B (I kappa B) kinase activation, I kappa B alpha degradation, and nuclear factor kappa B activation in HT-29 cells.Mol Pharmacol. 2004 Feb;65(2):342-9. doi: 10.1124/mol.65.2.342. Mol Pharmacol. 2004. PMID: 14742676
-
All three IkappaB isoforms and most Rel family members are stably associated with the IkappaB kinase 1/2 complex.Eur J Biochem. 1999 Jan;259(1-2):253-61. doi: 10.1046/j.1432-1327.1999.00028.x. Eur J Biochem. 1999. PMID: 9914500
-
Control of IkappaBalpha proteolysis by the ubiquitin-proteasome pathway.Biochimie. 2001 Mar-Apr;83(3-4):351-6. doi: 10.1016/s0300-9084(01)01237-8. Biochimie. 2001. PMID: 11295496 Review.
-
The role of nuclear factor-kappaB in the biology and treatment of multiple myeloma.Semin Oncol. 2001 Dec;28(6):626-33. doi: 10.1016/s0093-7754(01)90036-3. Semin Oncol. 2001. PMID: 11740821 Review.
Cited by
-
NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis.Proc Natl Acad Sci U S A. 2004 Jul 6;101(27):10137-42. doi: 10.1073/pnas.0403621101. Epub 2004 Jun 25. Proc Natl Acad Sci U S A. 2004. PMID: 15220474 Free PMC article.
-
Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway.Cardiovasc Res. 2010 Sep 1;87(4):656-64. doi: 10.1093/cvr/cvq148. Epub 2010 May 21. Cardiovasc Res. 2010. PMID: 20495188 Free PMC article.
-
The role of IKKβ in Venezuelan equine encephalitis virus infection.PLoS One. 2014 Feb 19;9(2):e86745. doi: 10.1371/journal.pone.0086745. eCollection 2014. PLoS One. 2014. PMID: 24586253 Free PMC article.
-
Nemo-like kinase (NLK) negatively regulates NF-kappa B activity through disrupting the interaction of TAK1 with IKKβ.Biochim Biophys Acta. 2014 Jul;1843(7):1365-72. doi: 10.1016/j.bbamcr.2014.03.028. Epub 2014 Apr 12. Biochim Biophys Acta. 2014. PMID: 24721172 Free PMC article.
-
Targeting the NFκB signaling pathways for breast cancer prevention and therapy.Curr Med Chem. 2015;22(2):264-89. doi: 10.2174/0929867321666141106124315. Curr Med Chem. 2015. PMID: 25386819 Free PMC article. Review.
References
-
- Baichwal, V. R., and P. A. Baeuerle. 1997. Activate NF-κB or die? Curr. Biol. 7:R94-R96. - PubMed
-
- Bonnard, M., C. Mirtsos, S. Suzuki, K. Graham, J. Huang, M. Ng, A. Itie, A. Wakeham, A. Shahinian, W. J. Henzel, A. J. Elia, W. Shillinglaw, T. W. Mak, Z. Cao, and W. C. Yeh. 2000. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-κB-dependent gene transcription. EMBO J. 19:4976-4985. - PMC - PubMed
-
- Brown, K., S. Gerstberger, L. Carlson, G. Franzoso, and U. Siebenlist. 1995. Control of IκB-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 267:1485-1488. - PubMed
-
- Chen, G., P. Cao, and D. V. Goeddel. 2002. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell 9:401-410. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous