

State of the art: why do the lungs of patients with cystic fibrosis become infected and why can't they clear the infection?
- PMID: 14511398
- PMCID: PMC203156
- DOI: 10.1186/1465-9921-4-8
State of the art: why do the lungs of patients with cystic fibrosis become infected and why can't they clear the infection?
Abstract
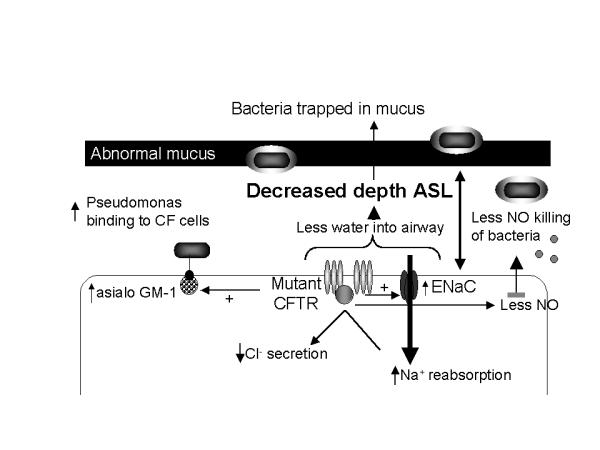
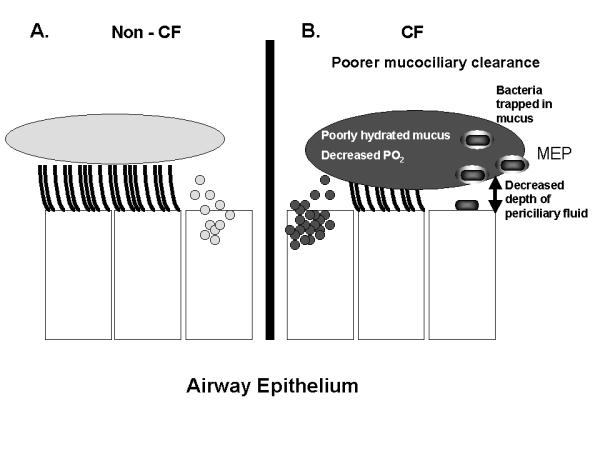
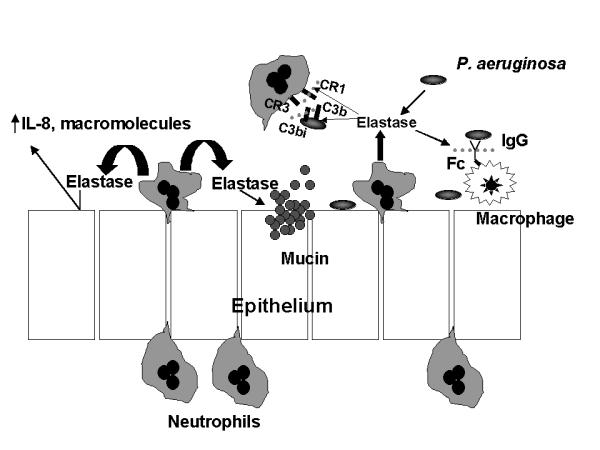
Cystic Fibrosis (CF) lung disease, which is characterized by airway obstruction, chronic bacterial infection, and an excessive inflammatory response, is responsible for most of the morbidity and mortality. Early in life, CF patients become infected with a limited spectrum of bacteria, especially P. aeruginosa. New data now indicate that decreased depth of periciliary fluid and abnormal hydration of mucus, which impede mucociliary clearance, contribute to initial infection. Diminished production of the antibacterial molecule nitric oxide, increased bacterial binding sites (e.g., asialo GM-1) on CF airway epithelial cells, and adaptations made by the bacteria to the airway microenvironment, including the production of virulence factors and the ability to organize into a biofilm, contribute to susceptibility to initial bacterial infection. Once the patient is infected, an overzealous inflammatory response in the CF lung likely contributes to the host's inability to eradicate infection. In response to increased IL-8 and leukotriene B4 production, neutrophils infiltrate the lung where they release mediators, such as elastase, that further inhibit host defenses, cripple opsonophagocytosis, impair mucociliary clearance, and damage airway wall architecture. The combination of these events favors the persistence of bacteria in the airway. Until a cure is discovered, further investigations into therapies that relieve obstruction, control infection, and attenuate inflammation offer the best hope of limiting damage to host tissues and prolonging survival.
Figures
Similar articles
-
Infection and inflammation in cystic fibrosis: a short review.J Cyst Fibros. 2005 Aug;4 Suppl 2:3-5. doi: 10.1016/j.jcf.2005.05.005. J Cyst Fibros. 2005. PMID: 15970469 Review.
-
Airway epithelial cell inflammatory signalling in cystic fibrosis.Int J Biochem Cell Biol. 2008;40(9):1703-15. doi: 10.1016/j.biocel.2008.02.002. Epub 2008 Feb 14. Int J Biochem Cell Biol. 2008. PMID: 18434235 Review.
-
Lower airway inflammation in infants with cystic fibrosis detected by newborn screening.Pediatr Pulmonol. 2005 Dec;40(6):500-10. doi: 10.1002/ppul.20294. Pediatr Pulmonol. 2005. PMID: 16208679
-
Acute Pseudomonas challenge in cystic fibrosis mice causes prolonged nuclear factor-kappa B activation, cytokine secretion, and persistent lung inflammation.J Allergy Clin Immunol. 2006 May;117(5):1163-9. doi: 10.1016/j.jaci.2006.01.052. J Allergy Clin Immunol. 2006. PMID: 16675347
-
Anti-inflammatory effect of augmented nitric oxide production in chronic lung infection.J Pathol. 2006 Jun;209(2):198-205. doi: 10.1002/path.1963. J Pathol. 2006. PMID: 16538611
Cited by
-
The Dynamics of Disease Progression in Cystic Fibrosis.PLoS One. 2016 Jun 1;11(6):e0156752. doi: 10.1371/journal.pone.0156752. eCollection 2016. PLoS One. 2016. PMID: 27248696 Free PMC article.
-
Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair.Am J Physiol Cell Physiol. 2010 Nov;299(5):C912-21. doi: 10.1152/ajpcell.00215.2010. Epub 2010 Aug 4. Am J Physiol Cell Physiol. 2010. PMID: 20686068 Free PMC article.
-
Lymphocytes in cystic fibrosis lung disease: a tale of two immunities.Clin Exp Immunol. 2004 Mar;135(3):358-60. doi: 10.1111/j.1365-2249.2003.02389.x. Clin Exp Immunol. 2004. PMID: 15008966 Free PMC article. No abstract available.
-
Assessment of hypoxia in children with cystic fibrosis.Arch Dis Child. 2005 Nov;90(11):1138-43. doi: 10.1136/adc.2005.071795. Arch Dis Child. 2005. PMID: 16243867 Free PMC article. Review.
-
Liquid movement across the surface epithelium of large airways.Respir Physiol Neurobiol. 2007 Dec 15;159(3):256-70. doi: 10.1016/j.resp.2007.06.005. Epub 2007 Jun 17. Respir Physiol Neurobiol. 2007. PMID: 17692578 Free PMC article. Review.
References
-
- Davis PB, Drumm ML, Konstan MW. State of the Art: Cystic Fibrosis. Am J Resp Crit Care Med. 1996;154:1229–1256. - PubMed
-
- Lloyd-Still JD. Crohn's disease and cystic fibrosis. Dig Dis Sci. 1994;39:880–885. - PubMed
-
- Taylor CJ, Aswani N. The pancreas in cystic fibrosis. Paediatr Respir Rev. 2002;3:77–81. - PubMed
-
- Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79(1 Suppl):S215–S255. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
