

Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo
- PMID: 12915549
- PMCID: PMC187414
- DOI: 10.1128/jvi.77.17.9337-9345.2003
Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo
Abstract
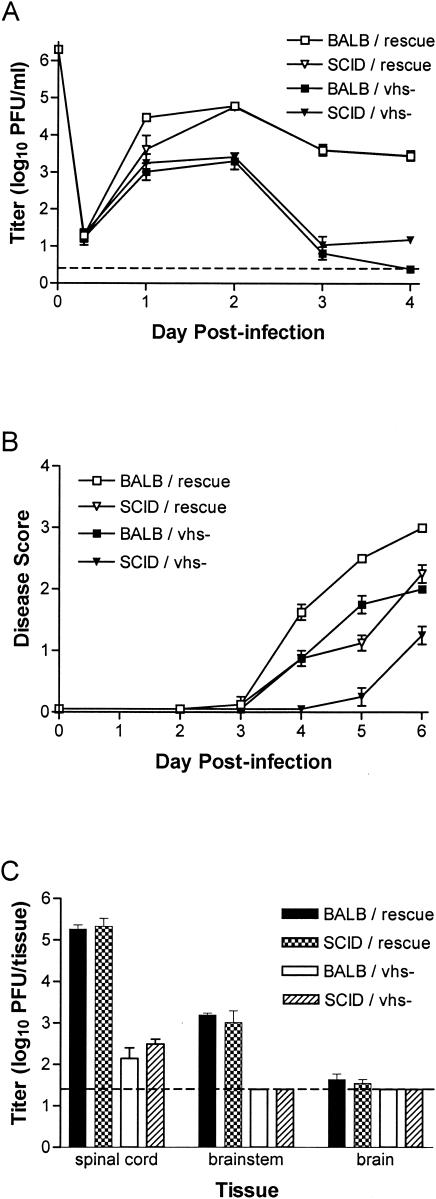
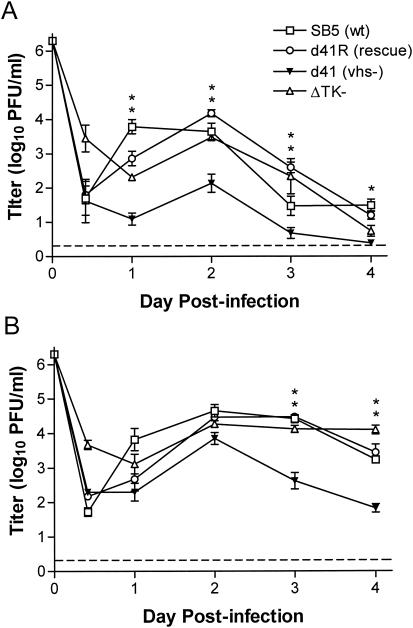
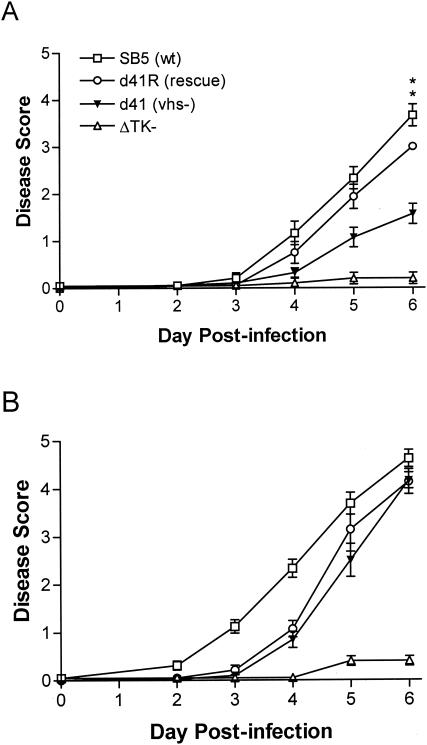
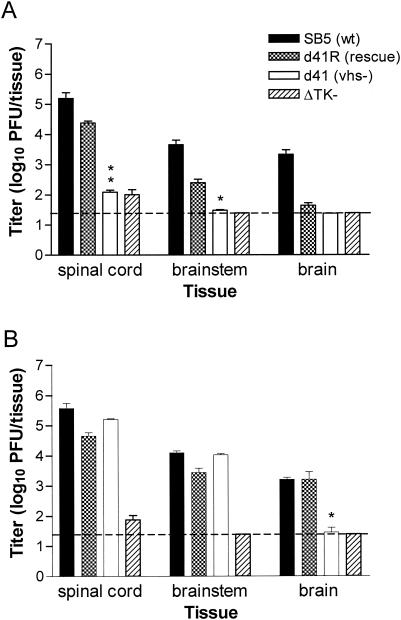
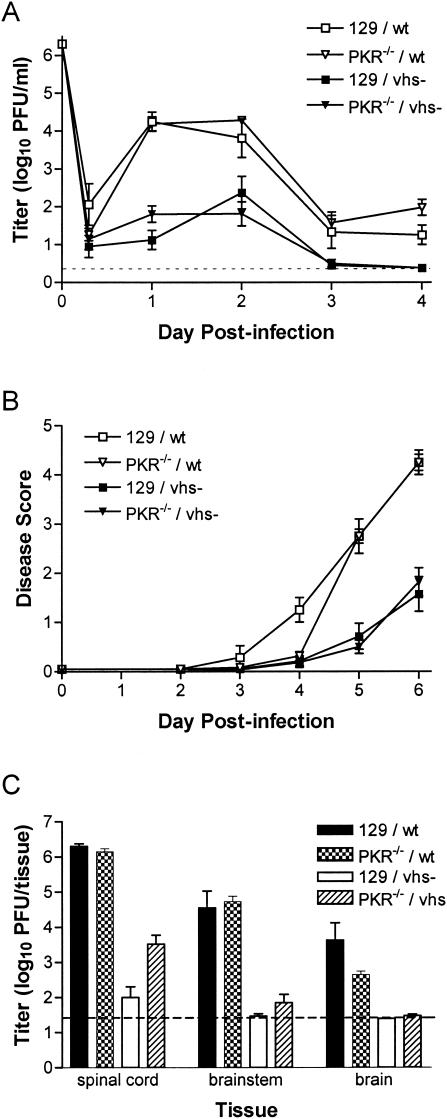
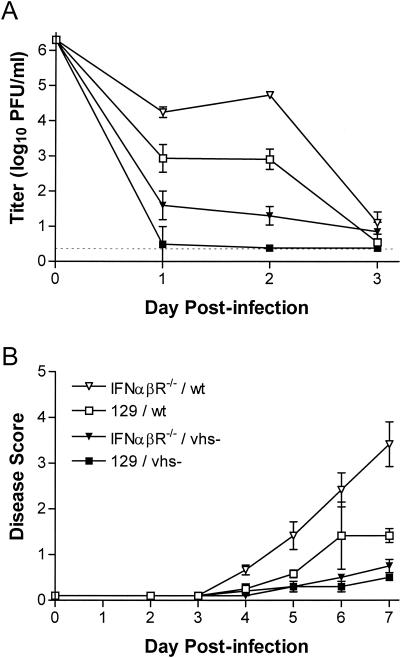
The herpes simplex virus (HSV) virion host shutoff (vhs) protein, the product of the UL41 (vhs) gene, is an important determinant of HSV virulence. vhs has been implicated in HSV interference with host antiviral immune responses, down-regulating expression of major histocompatibility complex molecules to help HSV evade host adaptive immunity. The severe attenuation of vhs-deficient viruses in vivo could reflect their inability to escape immune detection. To test this hypothesis, BALB/c or congenic SCID mice were infected intravaginally (i.vag.) with the HSV type 2 (HSV-2) vhs null mutant 333d41 or the vhs rescue virus 333d41(R). vhs-deficient virus remained severely attenuated in SCID mice compared with rescue virus, indicating that vhs regulation of adaptive immune responses does not influence HSV pathogenesis during acute infection. Innate antiviral effectors remain intact in SCID mice; prominent among these is alpha/beta interferon (IFN-alpha/beta). The attenuation of HSV-2 vhs mutants could reflect their failure to suppress IFN-alpha/beta-mediated antiviral activity. To test this hypothesis, 129 and congenic IFN-alpha/beta receptor-deficient (IFN-alpha/betaR(-/-)) mice were infected i.vag. with wild-type virus, vhs null mutants 333-vhsB or 333d41, or the vhs rescue virus 333d41(R). Whereas vhs-deficient viruses showed greatly reduced replication in the genital mucosa of 129 mice compared with wild-type or vhs rescue viruses, they were restored to nearly wild-type levels of replication in IFN-alpha/betaR(-/-) mice over the first 2 days postinfection. Only wild-type and vhs rescue viruses caused severe genital disease and hind limb paralysis in 129 mice, but infection of IFN-alpha/betaR(-/-) mice restored the virulence of vhs-deficient viruses. vhs-deficient viruses replicated as vigorously as wild-type and rescue viruses in the nervous systems of IFN-alpha/betaR(-/-) mice. Restoration was specific for the vhs mutation, because thymidine kinase-deficient HSV-2 did not regain virulence or the capacity to replicate in the nervous systems of IFN-alpha/betaR(-/-) mice. Furthermore, the defect in the IFN-alpha/beta response was required for restoration of vhs-deficient virus replication and virulence, but the IFN-alpha/beta-stimulated protein kinase R pathway was not involved. Finally, vhs of HSV-2 has a unique capacity to interfere with the IFN-alpha/beta response in vivo, because an HSV-1 vhs null mutant did not recover replication and virulence after i.vag. inoculation into IFN-alpha/betaR(-/-) mice. These results indicate that vhs plays an important role early in HSV-2 pathogenesis in vivo by interfering with the IFN-alpha/beta-mediated antiviral response.
Figures
Similar articles
-
Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness.Virology. 2004 Apr 25;322(1):158-67. doi: 10.1016/j.virol.2004.01.019. Virology. 2004. PMID: 15063125
-
Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo.J Exp Med. 1999 Feb 15;189(4):663-72. doi: 10.1084/jem.189.4.663. J Exp Med. 1999. PMID: 9989981 Free PMC article.
-
Selective ablation of virion host shutoff protein RNase activity attenuates herpes simplex virus 2 in mice.J Virol. 2008 Apr;82(7):3642-53. doi: 10.1128/JVI.02409-07. Epub 2008 Jan 30. J Virol. 2008. PMID: 18234805 Free PMC article.
-
Early shutoff of host protein synthesis in cells infected with herpes simplex viruses.Acta Virol. 2001;45(5-6):269-77. Acta Virol. 2001. PMID: 12083325 Review.
-
Innate immunity to herpes simplex virus type 2.Viral Immunol. 2003;16(4):475-90. doi: 10.1089/088282403771926300. Viral Immunol. 2003. PMID: 14733735 Review.
Cited by
-
mRNA decay during herpes simplex virus (HSV) infections: mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein.J Virol. 2013 Jan;87(1):94-109. doi: 10.1128/JVI.01557-12. Epub 2012 Oct 17. J Virol. 2013. PMID: 23077305 Free PMC article.
-
Use of transcriptional profiling to delineate the initial response of mice to intravaginal herpes simplex virus type 2 infection.Viral Immunol. 2013 Jun;26(3):172-9. doi: 10.1089/vim.2012.0093. Epub 2013 May 2. Viral Immunol. 2013. PMID: 23638732 Free PMC article.
-
The Herpes Simplex Virus Virion Host Shutoff Protein Enhances Translation of Viral True Late mRNAs Independently of Suppressing Protein Kinase R and Stress Granule Formation.J Virol. 2016 Jun 10;90(13):6049-6057. doi: 10.1128/JVI.03180-15. Print 2016 Jul 1. J Virol. 2016. PMID: 27099317 Free PMC article.
-
Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency.PLoS Pathog. 2011 Jul;7(7):e1002150. doi: 10.1371/journal.ppat.1002150. Epub 2011 Jul 21. PLoS Pathog. 2011. PMID: 21811408 Free PMC article.
-
The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-κB in dendritic cells in the absence of type I interferon signaling.J Virol. 2011 Dec;85(23):12662-72. doi: 10.1128/JVI.05557-11. Epub 2011 Sep 21. J Virol. 2011. PMID: 21937652 Free PMC article.
References
-
- Cassady, K. A., M. Gross, and B. Roizman. 1998. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the α subunit of eukaryotic translation initiation factor 2. J. Virol. 72:8620-8626. - PMC - PubMed
-
- Chou, J., J. J. Chen, M. Gross, and B. Roizman. 1995. Association of a Mr 90, 000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with γ134.5-mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 92:10516-10520. - PMC - PubMed
-
- Clements, G. B., and N. A. Stow. 1989. A herpes simplex virus type 1 mutant containing a deletion within immediate early gene 1 is latency-competent in mice. J. Gen. Virol. 70:2501-2506. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
