

Computational identification of protein coding potential of conserved sequence tags through cross-species evolutionary analysis
- PMID: 12888525
- PMCID: PMC169873
- DOI: 10.1093/nar/gkg483
Computational identification of protein coding potential of conserved sequence tags through cross-species evolutionary analysis
Abstract
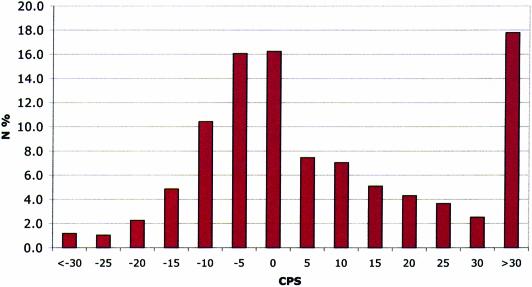
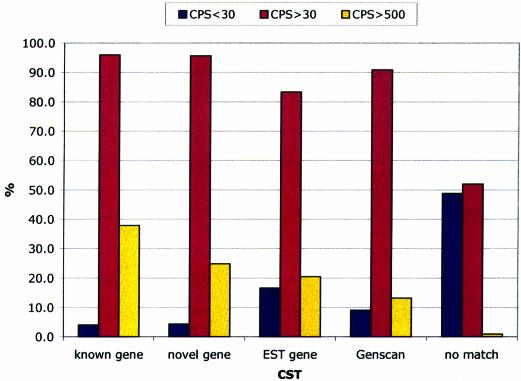
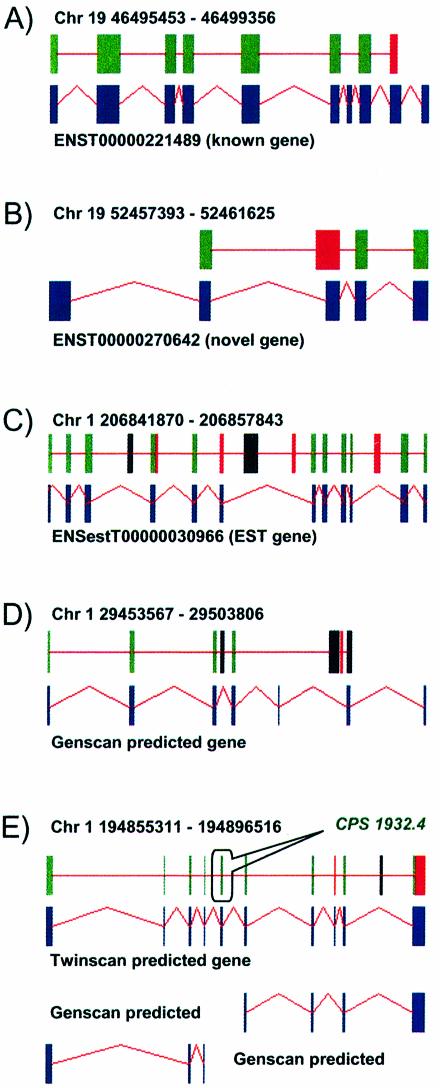
The identification of conserved sequence tags (CSTs) through comparative genome analysis may reveal important regulatory elements involved in shaping the spatio-temporal expression of genetic information. It is well known that the most significant fraction of CSTs observed in human-mouse comparisons correspond to protein coding exons, due to their strong evolutionary constraints. As we still do not know the complete gene inventory of the human and mouse genomes it is of the utmost importance to establish if detected conserved sequences are genes or not. We propose here a simple algorithm that, based on the observation of the specific evolutionary dynamics of coding sequences, efficiently discriminates between coding and non-coding CSTs. The application of this method may help the validation of predicted genes, the prediction of alternative splicing patterns in known and unknown genes and the definition of a dictionary of non-coding regulatory elements.
Figures
Similar articles
-
Improving the specificity of exon prediction using comparative genomics.BMC Genomics. 2008 Sep 16;9 Suppl 2(Suppl 2):S13. doi: 10.1186/1471-2164-9-S2-S13. BMC Genomics. 2008. PMID: 18831778 Free PMC article.
-
Genome-wide identification of coding and non-coding conserved sequence tags in human and mouse genomes.BMC Genomics. 2008 Jun 11;9:277. doi: 10.1186/1471-2164-9-277. BMC Genomics. 2008. PMID: 18547402 Free PMC article.
-
GenoMiner: a tool for genome-wide search of coding and non-coding conserved sequence tags.Bioinformatics. 2006 Feb 15;22(4):497-9. doi: 10.1093/bioinformatics/bti754. Epub 2005 Nov 2. Bioinformatics. 2006. PMID: 16267081
-
Comparative genomics as a tool for gene discovery.Curr Opin Biotechnol. 2006 Apr;17(2):161-7. doi: 10.1016/j.copbio.2006.01.007. Epub 2006 Feb 3. Curr Opin Biotechnol. 2006. PMID: 16459073 Review.
-
De novo prediction of structured RNAs from genomic sequences.Trends Biotechnol. 2010 Jan;28(1):9-19. doi: 10.1016/j.tibtech.2009.09.006. Epub 2009 Nov 26. Trends Biotechnol. 2010. PMID: 19942311 Free PMC article. Review.
Cited by
-
Molecular Functions of Long Non-Coding RNAs in Plants.Genes (Basel). 2012 Mar 8;3(1):176-90. doi: 10.3390/genes3010176. Genes (Basel). 2012. PMID: 24704849 Free PMC article.
-
Performance and scalability of discriminative metrics for comparative gene identification in 12 Drosophila genomes.PLoS Comput Biol. 2008 Apr 18;4(4):e1000067. doi: 10.1371/journal.pcbi.1000067. PLoS Comput Biol. 2008. PMID: 18421375 Free PMC article.
-
DG-CST (Disease Gene Conserved Sequence Tags), a database of human-mouse conserved elements associated to disease genes.Nucleic Acids Res. 2005 Jan 1;33(Database issue):D505-10. doi: 10.1093/nar/gki011. Nucleic Acids Res. 2005. PMID: 15608249 Free PMC article.
-
Statistical assessment of discriminative features for protein-coding and non coding cross-species conserved sequence elements.BMC Bioinformatics. 2009 Jun 16;10 Suppl 6(Suppl 6):S2. doi: 10.1186/1471-2105-10-S6-S2. BMC Bioinformatics. 2009. PMID: 19534745 Free PMC article.
-
Differentiating protein-coding and noncoding RNA: challenges and ambiguities.PLoS Comput Biol. 2008 Nov;4(11):e1000176. doi: 10.1371/journal.pcbi.1000176. Epub 2008 Nov 28. PLoS Comput Biol. 2008. PMID: 19043537 Free PMC article. Review.
References
-
- Burge C. and Karlin,S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol., 268, 78–94. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
