

A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein
- PMID: 12815074
- PMCID: PMC196135
- DOI: 10.1101/gad.260003
A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein
Abstract
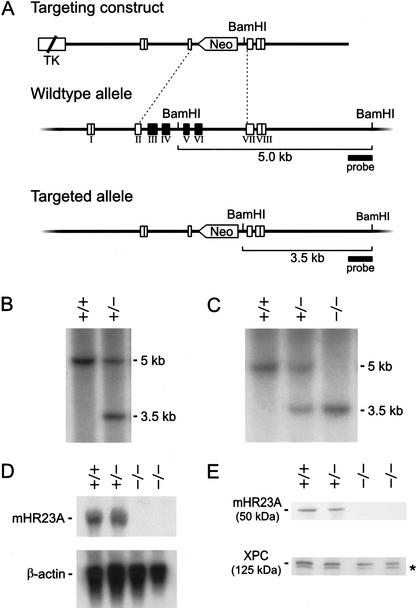
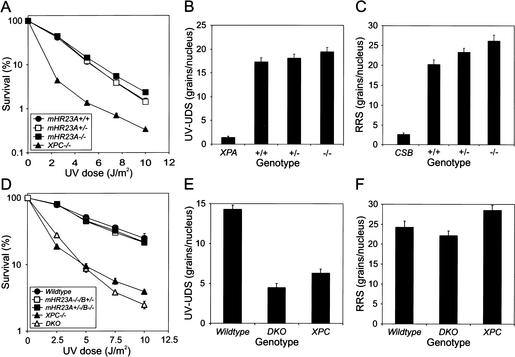
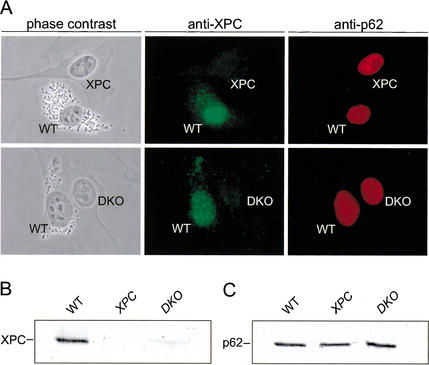
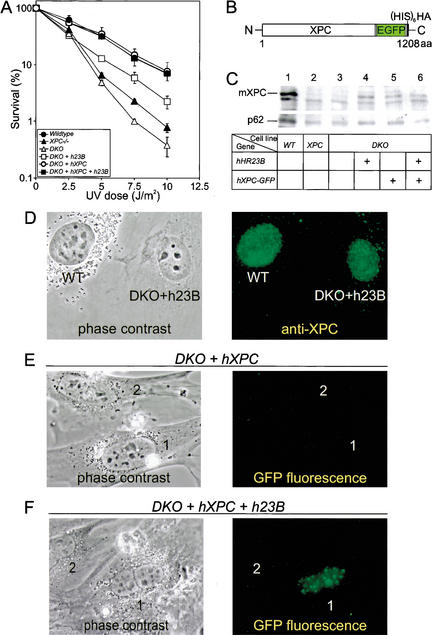
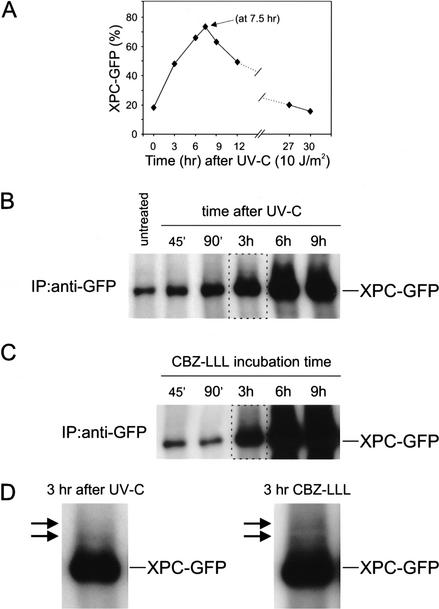
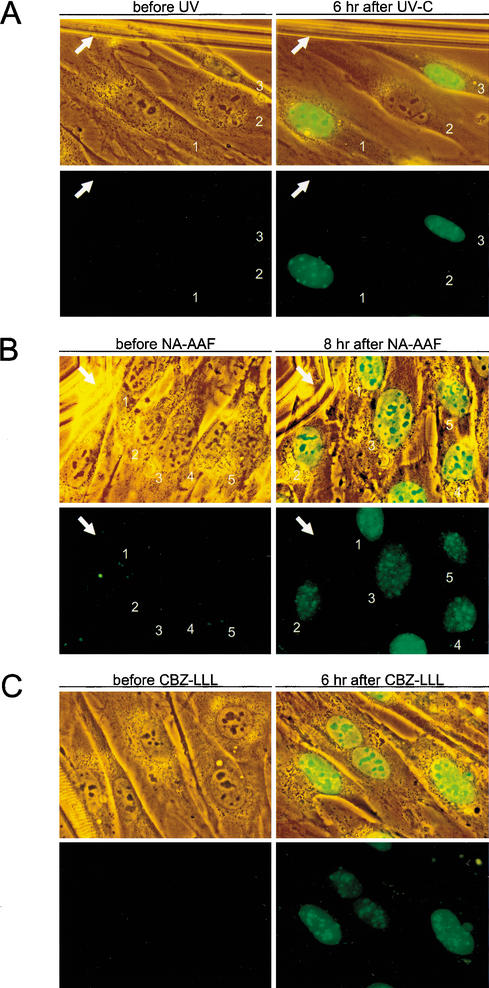
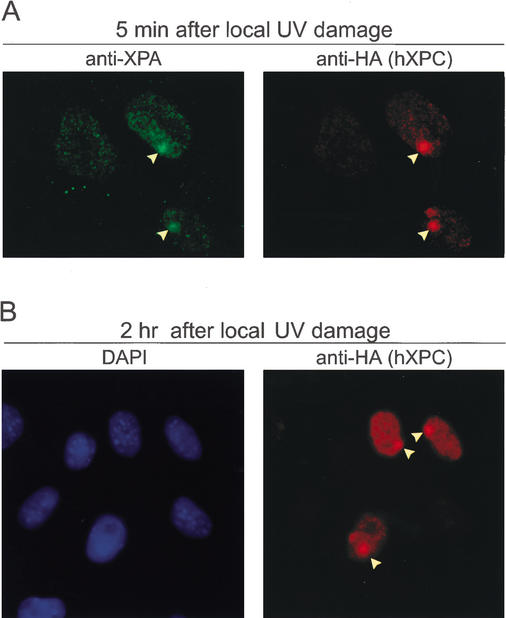
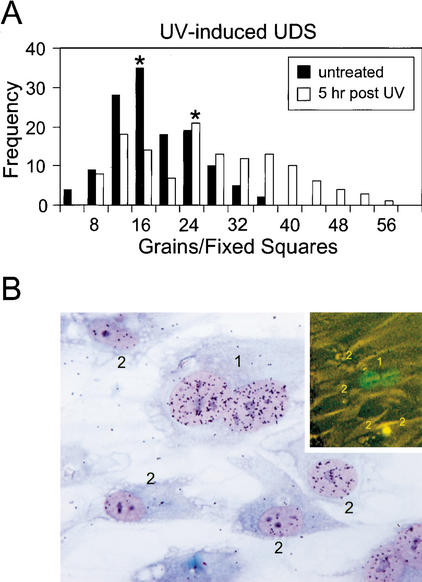
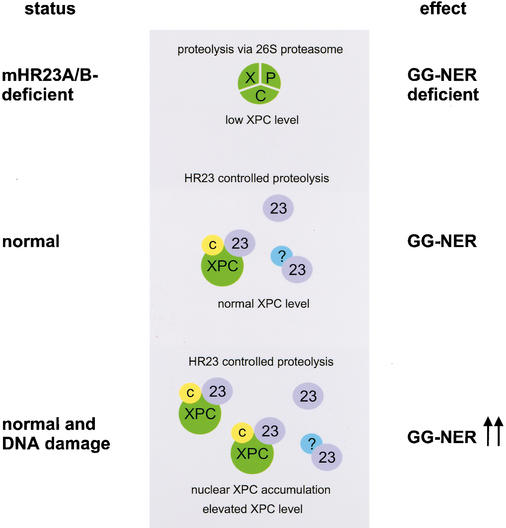
Primary DNA damage sensing in mammalian global genome nucleotide excision repair (GG-NER) is performed by the xeroderma pigmentosum group C (XPC)/HR23B protein complex. HR23B and HR23A are human homologs of the yeast ubiquitin-domain repair factor RAD23, the function of which is unknown. Knockout mice revealed that mHR23A and mHR23B have a fully redundant role in NER, and a partially redundant function in embryonic development. Inactivation of both genes causes embryonic lethality, but appeared still compatible with cellular viability. Analysis of mHR23A/B double-mutant cells showed that HR23 proteins function in NER by governing XPC stability via partial protection against proteasomal degradation. Interestingly, NER-type DNA damage further stabilizes XPC and thereby enhances repair. These findings resolve the primary function of RAD23 in repair and reveal a novel DNA-damage-dependent regulation mechanism of DNA repair in eukaryotes, which may be part of a more global damage-response circuitry.
Figures
Similar articles
-
Relative levels of the two mammalian Rad23 homologs determine composition and stability of the xeroderma pigmentosum group C protein complex.DNA Repair (Amst). 2004 Oct 5;3(10):1285-95. doi: 10.1016/j.dnarep.2004.06.010. DNA Repair (Amst). 2004. PMID: 15336624
-
Two mammalian homologs of yeast Rad23, HR23A and HR23B, as multifunctional proteins.Gene. 2017 Jan 15;597:1-9. doi: 10.1016/j.gene.2016.10.027. Epub 2016 Oct 20. Gene. 2017. PMID: 27771451 Review.
-
Developmental defects and male sterility in mice lacking the ubiquitin-like DNA repair gene mHR23B.Mol Cell Biol. 2002 Feb;22(4):1233-45. doi: 10.1128/MCB.22.4.1233-1245.2002. Mol Cell Biol. 2002. PMID: 11809813 Free PMC article.
-
Cloning, comparative mapping, and RNA expression of the mouse homologues of the Saccharomyces cerevisiae nucleotide excision repair gene RAD23.Genomics. 1996 Jan 1;31(1):20-7. doi: 10.1006/geno.1996.0004. Genomics. 1996. PMID: 8808275
-
Mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair.Enzymes. 2019;45:99-138. doi: 10.1016/bs.enz.2019.06.004. Epub 2019 Jul 8. Enzymes. 2019. PMID: 31627884 Review.
Cited by
-
Genetic variability of genes in NER pathway influences the treatment outcome of gastric cancer.Int J Clin Exp Pathol. 2015 May 1;8(5):5563-9. eCollection 2015. Int J Clin Exp Pathol. 2015. PMID: 26191265 Free PMC article.
-
In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation.Mol Cell Biol. 2007 Oct;27(19):6606-14. doi: 10.1128/MCB.02166-06. Epub 2007 Aug 6. Mol Cell Biol. 2007. PMID: 17682058 Free PMC article.
-
DNA Oxidation and Excision Repair Pathways.Int J Mol Sci. 2019 Dec 3;20(23):6092. doi: 10.3390/ijms20236092. Int J Mol Sci. 2019. PMID: 31816862 Free PMC article. Review.
-
Targeted gene therapy of xeroderma pigmentosum cells using meganuclease and TALEN™.PLoS One. 2013 Nov 13;8(11):e78678. doi: 10.1371/journal.pone.0078678. eCollection 2013. PLoS One. 2013. PMID: 24236034 Free PMC article.
-
Nucleotide excision repair deficient mouse models and neurological disease.DNA Repair (Amst). 2008 Jul 1;7(7):1180-9. doi: 10.1016/j.dnarep.2007.12.006. Epub 2008 Feb 12. DNA Repair (Amst). 2008. PMID: 18272436 Free PMC article. Review.
References
-
- Amundson S.A., Patterson, A., Do, K.T., and Fornace Jr., A.J. 2002. A nucleotide excision repair master-switch: p53 regulated coordinate induction of global genomic repair genes. Cancer Biol. Ther. 1: 145–149. - PubMed
-
- Araki M., Masutani, C., Takemura, M., Uchida, A., Sugasawa, K., Kondoh, J., Ohkuma, Y., and Hanaoka, F. 2001. Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J. Biol. Chem. 276: 18665–18672. - PubMed
-
- Bertolaet B.L., Clarke, D.J., Wolff, M., Watson, M.H., Henze, M., Divita, G., and Reed, S.I. 2001. UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat. Struct. Biol. 8: 417–422. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous