

Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection
- PMID: 12781537
- PMCID: PMC7140172
- DOI: 10.1016/s0140-6736(03)13414-9
Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection
Erratum in
- Lancet. 2003 May 24;361(9371):1832
Abstract
Background: The cause of severe acute respiratory syndrome (SARS) has been identified as a new coronavirus. Whole genome sequence analysis of various isolates might provide an indication of potential strain differences of this new virus. Moreover, mutation analysis will help to develop effective vaccines.
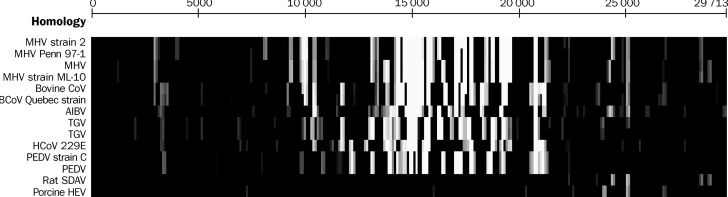
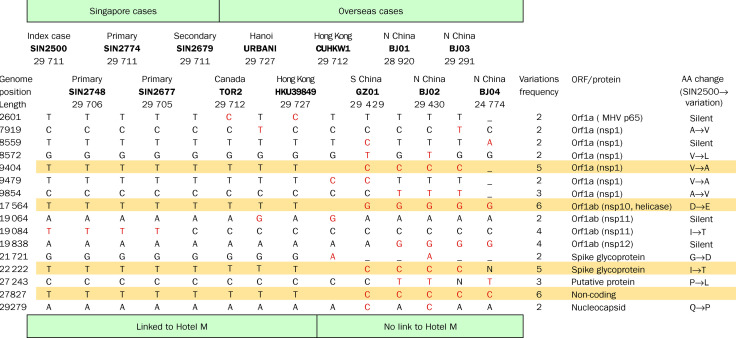
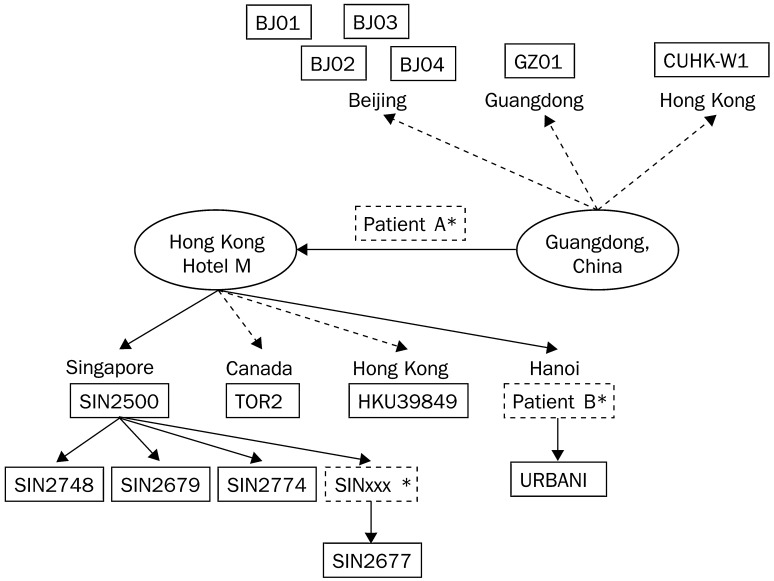
Methods: We sequenced the entire SARS viral genome of cultured isolates from the index case (SIN2500) presenting in Singapore, from three primary contacts (SIN2774, SIN2748, and SIN2677), and one secondary contact (SIN2679). These sequences were compared with the isolates from Canada (TOR2), Hong Kong (CUHK-W1 and HKU39849), Hanoi (URBANI), Guangzhou (GZ01), and Beijing (BJ01, BJ02, BJ03, BJ04).
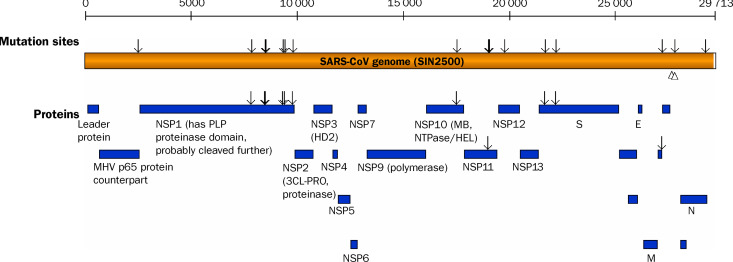
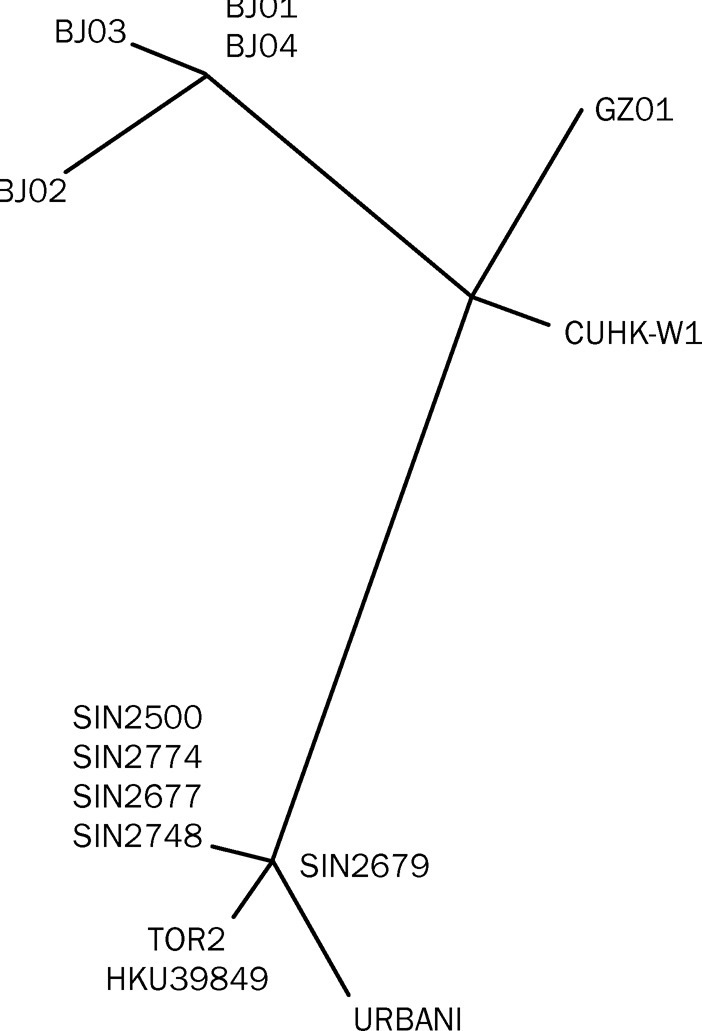
Findings: We identified 129 sequence variations among the 14 isolates, with 16 recurrent variant sequences. Common variant sequences at four loci define two distinct genotypes of the SARS virus. One genotype was linked with infections originating in Hotel M in Hong Kong, the second contained isolates from Hong Kong, Guangzhou, and Beijing with no association with Hotel M (p<0.0001). Moreover, other common sequence variants further distinguished the geographical origins of the isolates, especially between Singapore and Beijing.
Interpretation: Despite the recent onset of the SARS epidemic, genetic signatures are emerging that partition the worldwide SARS viral isolates into groups on the basis of contact source history and geography. These signatures can be used to trace sources of infection. In addition, a common variant associated with a non-conservative aminoacid change in the S1 region of the spike protein, suggests that immunological pressures might be starting to influence the evolution of the SARS virus in human populations.
Figures
Comment in
-
Comparative analysis of the SARS coronavirus genome: a good start to a long journey.Lancet. 2003 May 24;361(9371):1756-7. doi: 10.1016/S0140-6736(03)13444-7. Lancet. 2003. PMID: 12781529 Free PMC article. No abstract available.
-
Questions about comparative genomics of SARS coronavirus isolates.Lancet. 2003 Aug 16;362(9383):578; author reply 578-9. doi: 10.1016/S0140-6736(03)14130-X. Lancet. 2003. PMID: 12932399 Free PMC article. No abstract available.
Similar articles
-
Mutational dynamics of the SARS coronavirus in cell culture and human populations isolated in 2003.BMC Infect Dis. 2004 Sep 6;4:32. doi: 10.1186/1471-2334-4-32. BMC Infect Dis. 2004. PMID: 15347429 Free PMC article.
-
Complete genome sequences of the SARS-CoV: the BJ Group (Isolates BJ01-BJ04).Genomics Proteomics Bioinformatics. 2003 Aug;1(3):180-92. doi: 10.1016/s1672-0229(03)01023-4. Genomics Proteomics Bioinformatics. 2003. PMID: 15629030 Free PMC article.
-
Molecular biological analysis of genotyping and phylogeny of severe acute respiratory syndrome associated coronavirus.Chin Med J (Engl). 2004 Jan;117(1):42-8. Chin Med J (Engl). 2004. PMID: 14733771
-
The relationship of severe acute respiratory syndrome coronavirus with avian and other coronaviruses.Avian Dis. 2006 Sep;50(3):315-20. doi: 10.1637/7612-042006R.1. Avian Dis. 2006. PMID: 17039827 Review.
-
Bioinformatics research on the SARS coronavirus (SARS_CoV) in China.Curr Pharm Des. 2006;12(35):4565-72. doi: 10.2174/138161206779010404. Curr Pharm Des. 2006. PMID: 17168762 Review.
Cited by
-
SARS-CoV genome polymorphism: a bioinformatics study.Genomics Proteomics Bioinformatics. 2005 Feb;3(1):18-35. doi: 10.1016/s1672-0229(05)03004-4. Genomics Proteomics Bioinformatics. 2005. PMID: 16144519 Free PMC article.
-
The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain.FEBS J. 2006 Mar;273(5):1035-45. doi: 10.1111/j.1742-4658.2006.05130.x. FEBS J. 2006. PMID: 16478476 Free PMC article.
-
[Severe acute respiratory syndrome: a singular epidemic of viral pneumonia].Presse Med. 2004 Mar 13;33(5):344-51. doi: 10.1016/s0755-4982(04)98581-8. Presse Med. 2004. PMID: 15041887 Free PMC article. Review. French.
-
Comprehensive detection and identification of human coronaviruses, including the SARS-associated coronavirus, with a single RT-PCR assay.J Virol Methods. 2004 Dec 1;122(1):29-36. doi: 10.1016/j.jviromet.2004.07.008. J Virol Methods. 2004. PMID: 15488617 Free PMC article.
-
Severe acute respiratory syndrome, Beijing, 2003.Emerg Infect Dis. 2004 Jan;10(1):25-31. doi: 10.3201/eid1001.030553. Emerg Infect Dis. 2004. PMID: 15078593 Free PMC article.
References
-
- WHO Cumulative number of reported probable cases of severe acute respiratory syndrome (SARS) www.who.int/csr/sarscountry/2003_04_24/en/ (accessed April 19, 2003).
-
- Drosten C, Gunther S, Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003 (published online April 10, 10.1056/NEJMoa030781). - DOI - PubMed
-
- Ksiazek TG, Erdman D, Goldsmith CS. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003 (published online April 10, DOI: 10.1056/NEJMoa030747). - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
