

Human herpesvirus 8-encoded vGPCR activates nuclear factor of activated T cells and collaborates with human immunodeficiency virus type 1 Tat
- PMID: 12719569
- PMCID: PMC154031
- DOI: 10.1128/jvi.77.10.5759-5773.2003
Human herpesvirus 8-encoded vGPCR activates nuclear factor of activated T cells and collaborates with human immunodeficiency virus type 1 Tat
Abstract
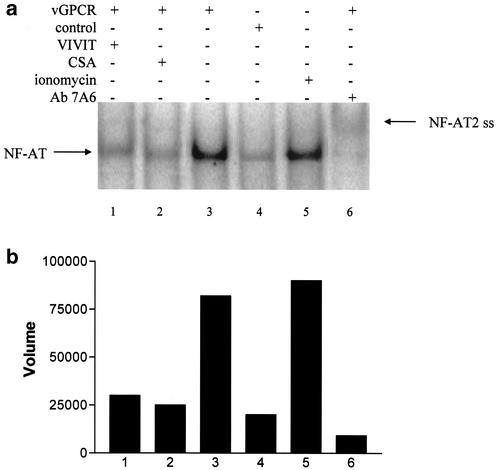
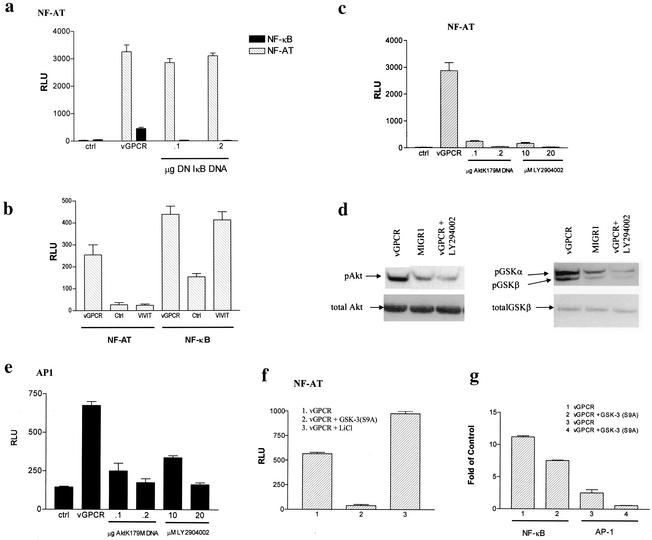
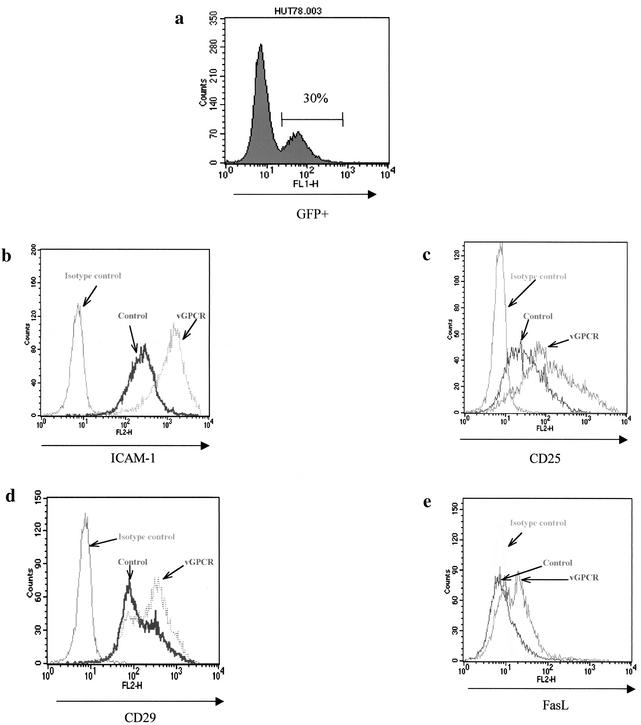
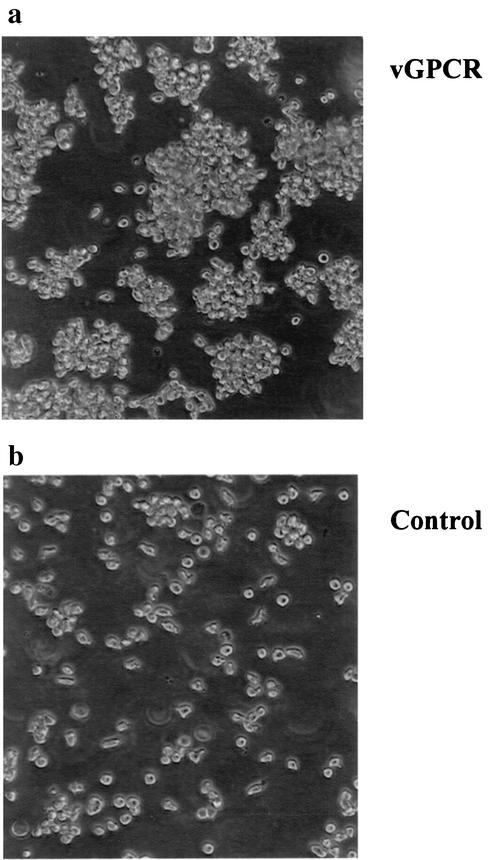
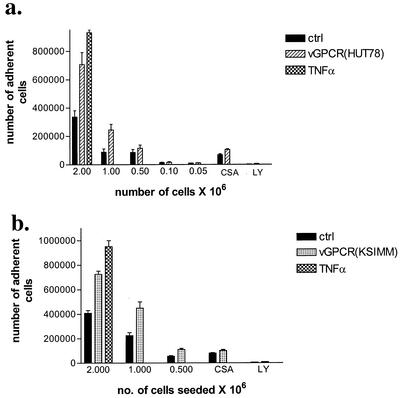
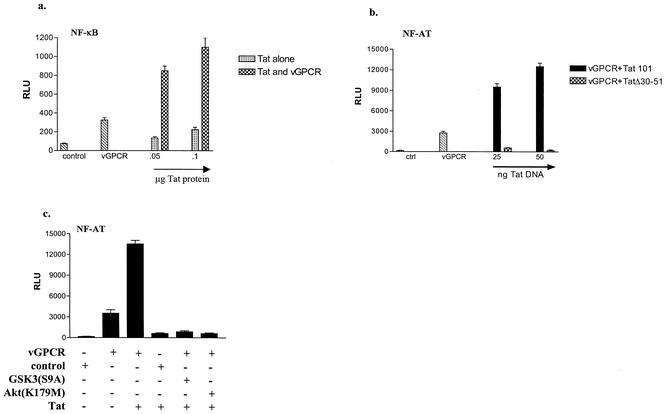
Human herpesvirus 8 (HHV-8), the etiologic agent of Kaposi's sarcoma (KS), encodes a chemokine receptor homologue, the viral G protein-coupled receptor (vGPCR), that has been implicated in KS pathogenesis. Expression of vGPCR constitutively activates several signaling pathways, including NF-kappa B, and induces the expression of proinflammatory and angiogenic factors, consistent with the inflammatory hyperproliferative nature of KS lesions. Here we show that vGPCR also constitutively activates the nuclear factor of activated T cells (NF-AT), another transcription factor important in regulation of the expression of inflammatory cytokines and related factors. NF-AT activation by vGPCR depended upon signaling through the phosphatidylinositol 3-kinase-Akt-glycogen synthetase kinase 3 (PI3-K/Akt/GSK-3) pathway and resulted in increased expression of NF-AT-dependent cell surface molecules (CD25, CD29, Fas ligand), proinflammatory cytokines (interleukin-2 [IL-2], IL-4), and proangiogenic factors (granulocyte-macrophage colony-stimulating factor GMCSF and TNF alpha). vGPCR expression also increased endothelial cell-T-cell adhesion. Although infection with HHV-8 is necessary to cause KS, coinfection with human immunodeficiency virus type 1 (HIV-1), in the absence of antiretroviral suppressive therapy, increases the risk of KS by many orders of magnitude. NF-AT and NF-kappa B activation by vGPCR was greatly increased by the HIV-1 Tat protein, although Tat alone had little effect on NF-AT. The enhancement of NF-AT by Tat appears to be mediated through collaborative stimulation of the PI3-K/Akt/GSK-3 pathway by vGPCR and Tat. Our data further support the idea that vGPCR contributes to the pathogenesis of KS by a paracrine mechanism and, in addition, provide the first evidence of collaboration between an HIV-1 protein and an HHV-8 protein.
Figures




Similar articles
-
Tumorigenesis by human herpesvirus 8 vGPCR is accelerated by human immunodeficiency virus type 1 Tat.J Virol. 2004 Sep;78(17):9336-42. doi: 10.1128/JVI.78.17.9336-9342.2004. J Virol. 2004. PMID: 15308728 Free PMC article.
-
Activation of NF-kappaB by the human herpesvirus 8 chemokine receptor ORF74: evidence for a paracrine model of Kaposi's sarcoma pathogenesis.J Virol. 2001 Sep;75(18):8660-73. doi: 10.1128/jvi.75.18.8660-8673.2001. J Virol. 2001. PMID: 11507211 Free PMC article.
-
The KSHV G protein-coupled receptor signals via multiple pathways to induce transcription factor activation in primary effusion lymphoma cells.Oncogene. 2004 Jan 15;23(2):514-23. doi: 10.1038/sj.onc.1207021. Oncogene. 2004. PMID: 14724579
-
Insights into the viral G protein-coupled receptor encoded by human herpesvirus type 8 (HHV-8).Biol Cell. 2004 Jun;96(5):349-54. doi: 10.1016/j.biolcel.2004.03.011. Biol Cell. 2004. PMID: 15207903 Review.
-
KSHV non-structural membrane proteins involved in the activation of intracellular signaling pathways and the pathogenesis of Kaposi's sarcoma.Curr Opin Virol. 2016 Oct;20:11-19. doi: 10.1016/j.coviro.2016.07.008. Epub 2016 Aug 9. Curr Opin Virol. 2016. PMID: 27518127 Review.
Cited by
-
Phosphoproteomic Analysis of KSHV-Infected Cells Reveals Roles of ORF45-Activated RSK during Lytic Replication.PLoS Pathog. 2015 Jul 2;11(7):e1004993. doi: 10.1371/journal.ppat.1004993. eCollection 2015 Jul. PLoS Pathog. 2015. PMID: 26133373 Free PMC article.
-
Kaposi's sarcoma associated herpesvirus G-protein coupled receptor activation of cyclooxygenase-2 in vascular endothelial cells.Virol J. 2007 Sep 14;4:87. doi: 10.1186/1743-422X-4-87. Virol J. 2007. PMID: 17868457 Free PMC article.
-
Herpesvirus-encoded GPCRs: neglected players in inflammatory and proliferative diseases?Nat Rev Drug Discov. 2014 Feb;13(2):123-39. doi: 10.1038/nrd4189. Epub 2014 Jan 21. Nat Rev Drug Discov. 2014. PMID: 24445563 Review.
-
Sulfated polymannuroguluronate inhibits Tat-induced SLK cell adhesion via a novel binding site, a KKR spatial triad.Acta Pharmacol Sin. 2011 May;32(5):647-54. doi: 10.1038/aps.2011.2. Epub 2011 Apr 18. Acta Pharmacol Sin. 2011. PMID: 21499289 Free PMC article.
-
Human immunodeficiency virus type 1 Tat accelerates Kaposi sarcoma-associated herpesvirus Kaposin A-mediated tumorigenesis of transformed fibroblasts in vitro as well as in nude and immunocompetent mice.Neoplasia. 2009 Dec;11(12):1272-84. doi: 10.1593/neo.09494. Neoplasia. 2009. PMID: 20019835 Free PMC article.
References
-
- Agwale, S. M., M. T. Shata, M. S. Reitz, Jr., V. S. Kalyanaraman, R. C. Gallo, M. Popovic, and D. M. Hone. 2002. A Tat subunit vaccine confers protective immunity against the immune-modulating activity of the human immunodeficiency virus type 1 Tat protein in mice. Proc. Natl. Acad. Sci. USA 99:10037-10041. - PMC - PubMed
-
- Albini, A., I. Paglieri, G. Orengo, S. Carlone, M. G. Aluigi, R. DeMarchi, C. Matteucci, A. Mantovani, F. Carozzi, S. Donini, and R. Benelli. 1997. The β-core fragment of human chorionic gonadotropin inhibits growth of Kaposi's sarcoma-derived cells and a new immortalized Kaposi's sarcoma cell line. AIDS 11:713-721. - PubMed
-
- Aramburu, J., M. B. Yaffe, C. Lopez-Rodriguez, L. C. Cantley, P. G. Hogan, and A. Rao. 1999. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285:2129-2133. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
