

The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury
- PMID: 12588885
- PMCID: PMC151924
- DOI: 10.1172/JCI16491
The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury
Abstract
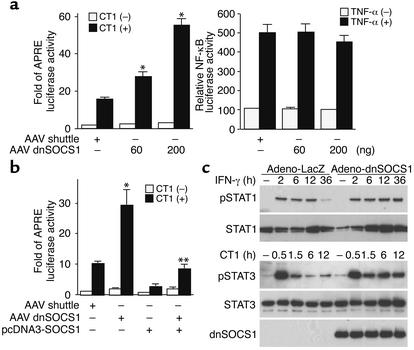
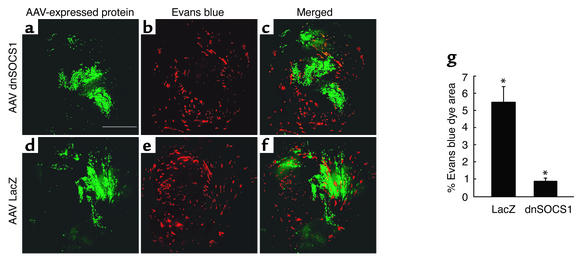
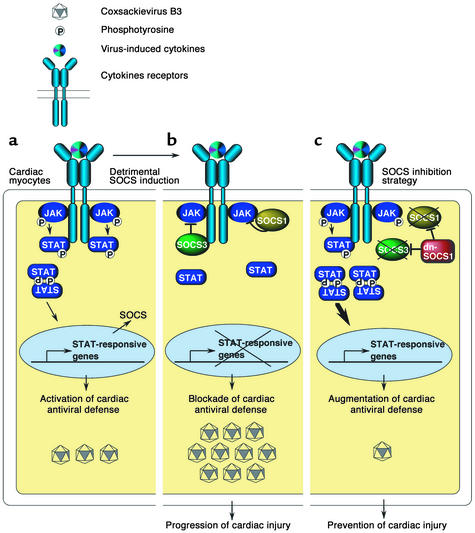
Enteroviral infections of the heart are among the most commonly identified causes of acute myocarditis in children and adults and have been implicated in dilated cardiomyopathy. Although there is considerable information regarding the cellular immune response in myocarditis, little is known about innate signaling mechanisms within the infected cardiac myocyte that contribute to the host defense against viral infection. Here we show the essential role of Janus kinase (JAK) signaling in cardiac myocyte antiviral defense and a negative role of an intrinsic JAK inhibitor, the suppressor of cytokine signaling (SOCS), in the early disease process. Cardiac myocyte-specific transgenic expression of SOCS1 inhibited enterovirus-induced signaling of JAK and the signal transducers and activators of transcription (STAT), with accompanying increases in viral replication, cardiomyopathy, and mortality in coxsackievirus-infected mice. Furthermore, the inhibition of SOCS in the cardiac myocyte through adeno-associated virus-mediated (AAV-mediated) expression of a dominant-negative SOCS1 increased the myocyte resistance to the acute cardiac injury caused by enteroviral infection. These results indicate that strategies directed at inhibition of SOCS in the heart and perhaps other organs can augment the host-cell antiviral system, thus preventing viral-mediated end-organ damage during the early stages of infection.
Figures
Similar articles
-
Innate defense mechanism against virus infection within the cardiac myocyte requiring gp130-STAT3 signaling.Circulation. 2006 Nov 28;114(22):2364-73. doi: 10.1161/CIRCULATIONAHA.106.642454. Epub 2006 Nov 13. Circulation. 2006. PMID: 17101849
-
The JAK-inhibitor, JAB/SOCS-1 selectively inhibits cytokine-induced, but not v-Src induced JAK-STAT activation.Oncogene. 2000 Sep 28;19(41):4795-801. doi: 10.1038/sj.onc.1203829. Oncogene. 2000. PMID: 11032030
-
Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS.J Clin Invest. 2002 Nov;110(10):1503-13. doi: 10.1172/JCI15841. J Clin Invest. 2002. PMID: 12438448 Free PMC article.
-
Unsolved medical issues and new targets for further research in viral myocarditis and dilated cardiomyopathy.Ernst Schering Res Found Workshop. 2006;(55):19-35. doi: 10.1007/3-540-30822-9_2. Ernst Schering Res Found Workshop. 2006. PMID: 16329655 Review.
-
Suppressors of cytokine signaling (SOCS): inhibitors of the JAK/STAT pathway.Shock. 2002 Feb;17(2):83-90. doi: 10.1097/00024382-200202000-00001. Shock. 2002. PMID: 11837794 Review.
Cited by
-
A respiratory syncytial virus persistent-infected cell line system reveals the involvement of SOCS1 in the innate antiviral response.Virol Sin. 2015 Jun;30(3):190-9. doi: 10.1007/s12250-015-3597-0. Epub 2015 Jun 19. Virol Sin. 2015. PMID: 26122642 Free PMC article.
-
SOCS proteins in infectious diseases of mammals.Vet Immunol Immunopathol. 2013 Jan 15;151(1-2):1-19. doi: 10.1016/j.vetimm.2012.11.008. Epub 2012 Nov 20. Vet Immunol Immunopathol. 2013. PMID: 23219158 Free PMC article. Review.
-
p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload.Mol Cell Biol. 2004 Dec;24(24):10611-20. doi: 10.1128/MCB.24.24.10611-10620.2004. Mol Cell Biol. 2004. PMID: 15572667 Free PMC article.
-
In vivo ablation of type I interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial disease.J Virol. 2014 May;88(9):5087-99. doi: 10.1128/JVI.00184-14. Epub 2014 Feb 26. J Virol. 2014. PMID: 24574394 Free PMC article.
-
Expression of SOCS1 and SOCS3 genes in human graft-versus-host disease after allogeneic hematopoietic stem cell transplantation.Blood Res. 2013 Mar;48(1):16-23. doi: 10.5045/br.2013.48.1.16. Epub 2013 Mar 25. Blood Res. 2013. PMID: 23589790 Free PMC article.
References
-
- Rose NR. Viral damage or “molecular mimicry” — placing the blame in myocarditis. Nat. Med. 2000;6:631–632. - PubMed
-
- Liu P, Martino T, Opavsky MA, Penninger J. Viral myocarditis: balance between viral infection and immune response. Can. J. Cardiol. 1996;12:935–943. - PubMed
-
- Baboonian C, Davies MJ, Booth JC, McKenna WJ. Coxsackie B viruses and human heart disease. Curr. Top. Microbiol. Immunol. 1997;223:31–52. - PubMed
-
- Badorff C, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 1999;5:320–326. - PubMed
-
- Knowlton KU, Badorff C. The immune system in viral myocarditis: maintaining the balance. Circ. Res. 1999;85:559–561. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
