

Accurate and automated classification of protein secondary structure with PsiCSI
- PMID: 12538892
- PMCID: PMC2312422
- DOI: 10.1110/ps.0222303
Accurate and automated classification of protein secondary structure with PsiCSI
Abstract
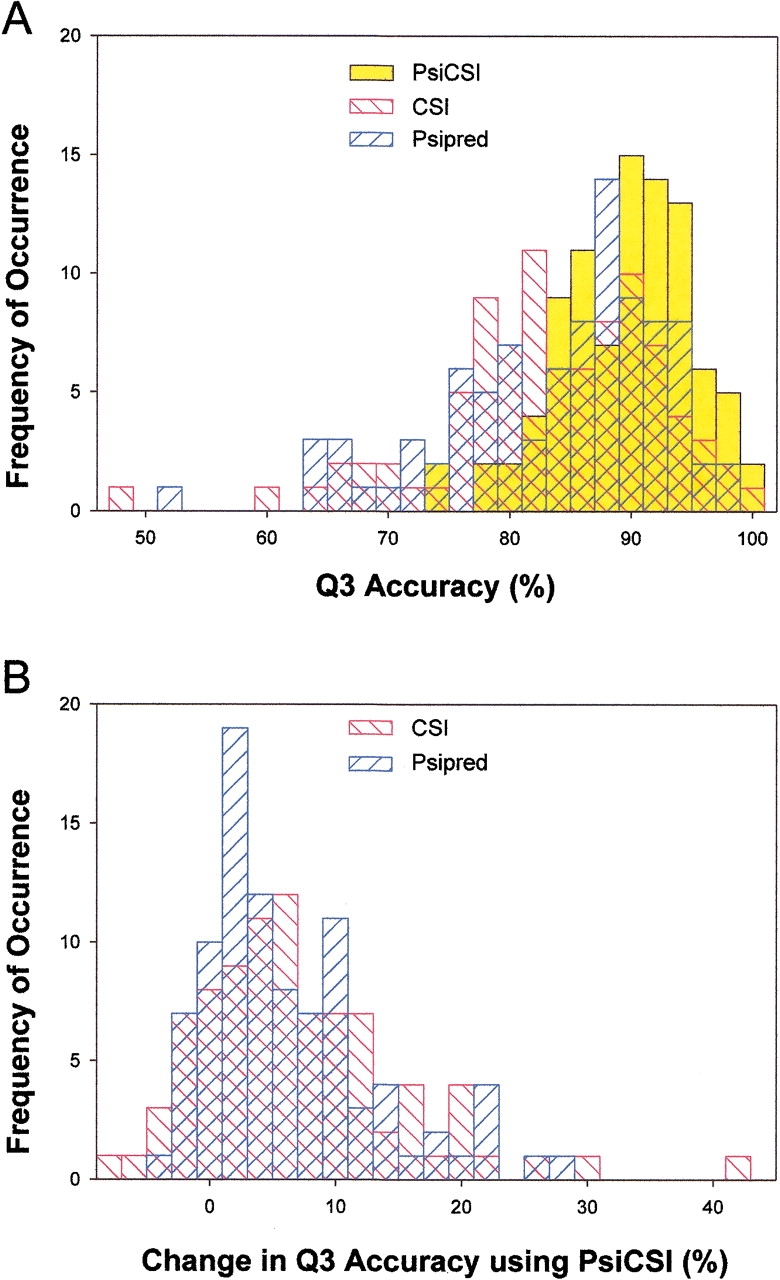
PsiCSI is a highly accurate and automated method of assigning secondary structure from NMR data, which is a useful intermediate step in the determination of tertiary structures. The method combines information from chemical shifts and protein sequence using three layers of neural networks. Training and testing was performed on a suite of 92 proteins (9437 residues) with known secondary and tertiary structure. Using a stringent cross-validation procedure in which the target and homologous proteins were removed from the databases used for training the neural networks, an average 89% Q3 accuracy (per residue) was observed. This is an increase of 6.2% and 5.5% (representing 36% and 33% fewer errors) over methods that use chemical shifts (CSI) or sequence information (Psipred) alone. In addition, PsiCSI improves upon the translation of chemical shift information to secondary structure (Q3 = 87.4%) and is able to use sequence information as an effective substitute for sparse NMR data (Q3 = 86.9% without (13)C shifts and Q3 = 86.8% with only H(alpha) shifts available). Finally, errors made by PsiCSI almost exclusively involve the interchange of helix or strand with coil and not helix with strand (<2.5 occurrences per 10000 residues). The automation, increased accuracy, absence of gross errors, and robustness with regards to sparse data make PsiCSI ideal for high-throughput applications, and should improve the effectiveness of hybrid NMR/de novo structure determination methods. A Web server is available for users to submit data and have the assignment returned.
Figures
Similar articles
-
Protein secondary structure prediction with SPARROW.J Chem Inf Model. 2012 Feb 27;52(2):545-56. doi: 10.1021/ci200321u. Epub 2012 Jan 23. J Chem Inf Model. 2012. PMID: 22224407
-
Combining prediction of secondary structure and solvent accessibility in proteins.Proteins. 2005 May 15;59(3):467-75. doi: 10.1002/prot.20441. Proteins. 2005. PMID: 15768403
-
A neural network method for prediction of beta-turn types in proteins using evolutionary information.Bioinformatics. 2004 Nov 1;20(16):2751-8. doi: 10.1093/bioinformatics/bth322. Epub 2004 May 14. Bioinformatics. 2004. PMID: 15145798
-
Automation of NMR structure determination of proteins.Curr Opin Struct Biol. 2004 Oct;14(5):547-53. doi: 10.1016/j.sbi.2004.09.003. Curr Opin Struct Biol. 2004. PMID: 15465314 Review.
-
Automated analysis of NMR assignments and structures for proteins.Curr Opin Struct Biol. 1999 Oct;9(5):635-42. doi: 10.1016/s0959-440x(99)00019-6. Curr Opin Struct Biol. 1999. PMID: 10508776 Review.
Cited by
-
Use of secondary structural information and C alpha-C alpha distance restraints to model protein structures with MODELLER.J Biosci. 2007 Aug;32(5):929-36. doi: 10.1007/s12038-007-0093-1. J Biosci. 2007. PMID: 17914235
-
PROTINFO: new algorithms for enhanced protein structure predictions.Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W77-80. doi: 10.1093/nar/gki403. Nucleic Acids Res. 2005. PMID: 15980581 Free PMC article.
-
TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts.J Biomol NMR. 2009 Aug;44(4):213-23. doi: 10.1007/s10858-009-9333-z. Epub 2009 Jun 23. J Biomol NMR. 2009. PMID: 19548092 Free PMC article.
-
PROTINFO: Secondary and tertiary protein structure prediction.Nucleic Acids Res. 2003 Jul 1;31(13):3296-9. doi: 10.1093/nar/gkg541. Nucleic Acids Res. 2003. PMID: 12824311 Free PMC article.
-
Uncovering symmetry-breaking vector and reliability order for assigning secondary structures of proteins from atomic NMR chemical shifts in amino acids.J Biomol NMR. 2011 Dec;51(4):411-24. doi: 10.1007/s10858-011-9579-0. Epub 2011 Oct 30. J Biomol NMR. 2011. PMID: 22038647
References
-
- Bailey-Kellogg, C., Widge, A., Kelley, J.J., Berardi, M.J., Bushweller, J.H., and Donald, B.R. 2000. The NOESY jigsaw: Automated protein secondary structure and main-chain assignment from sparse, unassigned NMR data. J. Comput. Biol. 7 537–558. - PubMed
-
- Bonneau, R., Tsai, J., Ruczinski, I., Chivian, D., Rohl, C., Strauss, C.E., and Baker, D. 2001. Rosetta in CASP4: Progress in ab initio protein structure prediction. Proteins 45 119–126. - PubMed
-
- Bonvin, A.M., Houben, K., Guenneugues, M., Kaptein, R., and Boelens, R. 2001. Rapid protein fold determination using secondary chemical shifts and cross-hydrogen bond 15N-13C′ scalar couplings (3hbJNC′). J. Biomol. NMR 21 221–233. - PubMed
-
- Brenner, S.E. 2001. A tour of structural genomics. Nat. Rev. Genet. 2 801–809. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
