

The PAAD/PYRIN-family protein ASC is a dual regulator of a conserved step in nuclear factor kappaB activation pathways
- PMID: 12486103
- PMCID: PMC2196065
- DOI: 10.1084/jem.20021552
The PAAD/PYRIN-family protein ASC is a dual regulator of a conserved step in nuclear factor kappaB activation pathways
Abstract
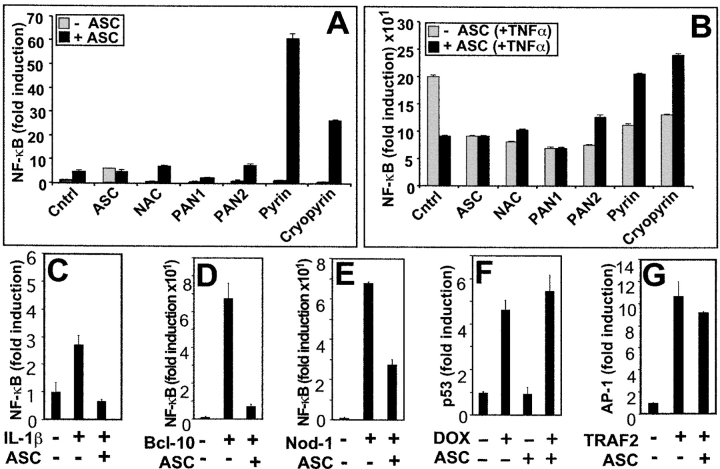
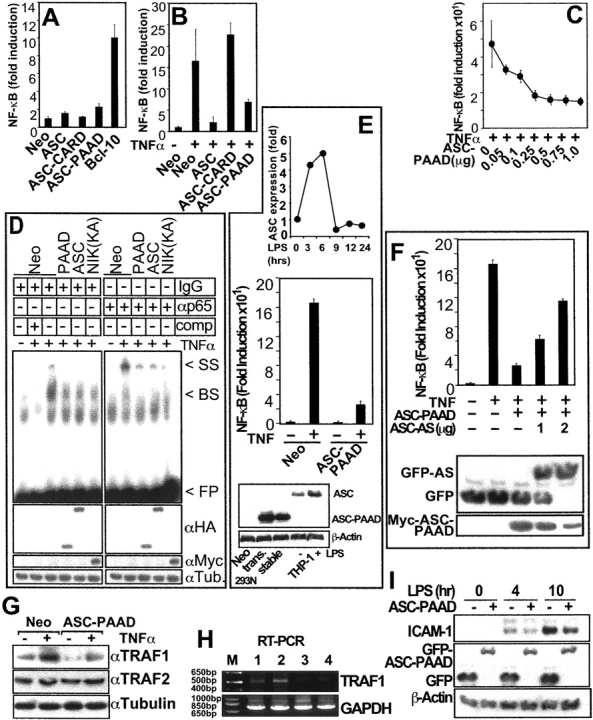
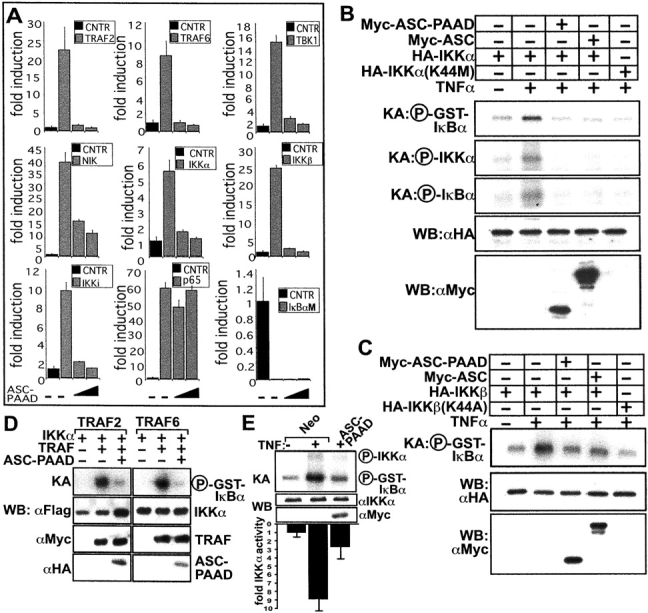
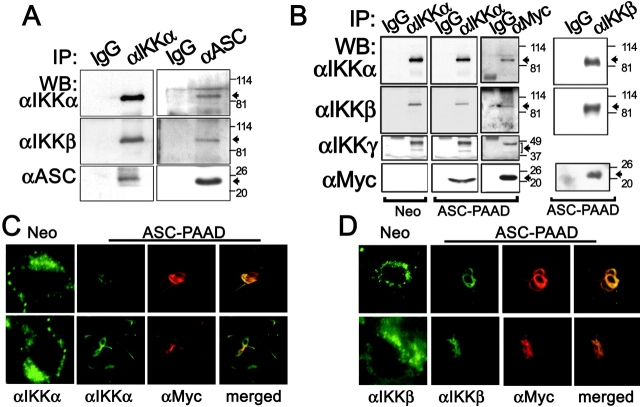
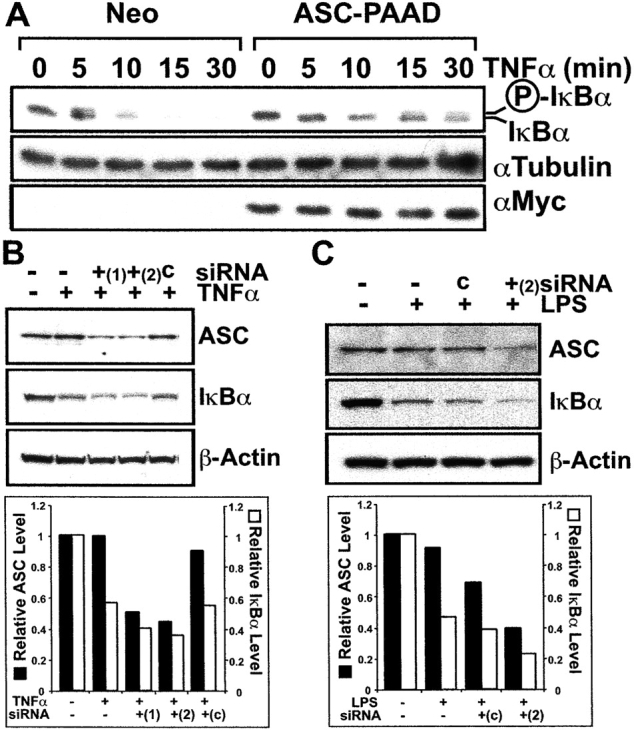
Apoptosis-associated speck-like protein containing a Caspase recruitment domain (ASC) belongs to a large family of proteins that contain a Pyrin, AIM, ASC, and death domain-like (PAAD) domain (also known as PYRIN, DAPIN, Pyk). Recent data have suggested that ASC functions as an adaptor protein linking various PAAD-family proteins to pathways involved in nuclear factor (NF)-kappaB and pro-Caspase-1 activation. We present evidence here that the role of ASC in modulating NF-kappaB activation pathways is much broader than previously suspected, as it can either inhibit or activate NF-kappaB, depending on cellular context. While coexpression of ASC with certain PAAD-family proteins such as Pyrin and Cryopyrin increases NF-kappaB activity, ASC has an inhibitory influence on NF-kappaB activation by various proinflammatory stimuli, including tumor necrosis factor (TNF)alpha, interleukin 1beta, and lipopolysaccharide (LPS). Elevations in ASC protein levels or of the PAAD domain of ASC suppressed activation of IkappaB kinases in cells exposed to pro-inflammatory stimuli. Conversely, reducing endogenous levels of ASC using siRNA enhanced TNF- and LPS-induced degradation of the IKK substrate, IkappaBalpha. Our findings suggest that ASC modulates diverse NF-kappaB induction pathways by acting upon the IKK complex, implying a broad role for this and similar proteins containing PAAD domains in regulation of inflammatory responses.
Figures
Similar articles
-
Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation.J Immunol. 2003 Dec 1;171(11):6154-63. doi: 10.4049/jimmunol.171.11.6154. J Immunol. 2003. PMID: 14634131
-
The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation.Biochem J. 2003 Jul 1;373(Pt 1):101-13. doi: 10.1042/BJ20030304. Biochem J. 2003. PMID: 12656673 Free PMC article.
-
PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages.J Biol Chem. 2004 Dec 10;279(50):51897-907. doi: 10.1074/jbc.M406741200. Epub 2004 Sep 28. J Biol Chem. 2004. PMID: 15456791
-
Genetic and Epigenetic Regulation of the Innate Immune Response to Gout.Immunol Invest. 2023 Apr;52(3):364-397. doi: 10.1080/08820139.2023.2168554. Epub 2023 Feb 6. Immunol Invest. 2023. PMID: 36745138 Review.
-
Regulation and function of IKK and IKK-related kinases.Sci STKE. 2006 Oct 17;2006(357):re13. doi: 10.1126/stke.3572006re13. Sci STKE. 2006. PMID: 17047224 Review.
Cited by
-
High Frequency of Inherited Variants in the MEFV Gene in Acute Lymphocytic Leukemia.Indian J Hematol Blood Transfus. 2011 Sep;27(3):164-8. doi: 10.1007/s12288-011-0095-x. Epub 2011 Jul 21. Indian J Hematol Blood Transfus. 2011. PMID: 22942567 Free PMC article.
-
Expression and regulation of cryopyrin and related proteins in rheumatoid arthritis synovium.Ann Rheum Dis. 2005 May;64(5):708-14. doi: 10.1136/ard.2004.025577. Epub 2004 Oct 21. Ann Rheum Dis. 2005. PMID: 15498798 Free PMC article.
-
Grouper RIP2 inhibits Singapore grouper iridovirus infection by modulating ASC-caspase-1 interaction.Front Immunol. 2023 May 8;14:1185907. doi: 10.3389/fimmu.2023.1185907. eCollection 2023. Front Immunol. 2023. PMID: 37223098 Free PMC article.
-
COPs and POPs: modulators of inflammasome activity.J Immunol. 2007 Dec 15;179(12):7993-8. doi: 10.4049/jimmunol.179.12.7993. J Immunol. 2007. PMID: 18056338 Free PMC article. Review.
-
Curcumin: a double hit on malignant mesothelioma.Cancer Prev Res (Phila). 2014 Mar;7(3):330-40. doi: 10.1158/1940-6207.CAPR-13-0259. Epub 2014 Jan 15. Cancer Prev Res (Phila). 2014. PMID: 24431405 Free PMC article.
References
-
- Aravind, L., V.M. Dixit, and E.V. Koonin. 1999. The domains of death: evolution of the apoptosis machinery. Trends Biochem. Sci. 24:47–53. - PubMed
-
- Weber, C.H., and C. Vincenz. 2001. The death domain superfamily: a tale of two interfaces? Trends Biochem. Sci. 26:475–481. - PubMed
-
- Pawlowski, K., F. Pio, Z.-L. Chu, J.C. Reed, and A. Godzik. 2001. PAAD-a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem. Sci. 26:85–87. - PubMed
-
- Bertin, J., and P.S. DiStefano. 2000. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death Differ. 7:1273–1274. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
