

Role of protein kinase C betaII in influenza virus entry via late endosomes
- PMID: 12477851
- PMCID: PMC140583
- DOI: 10.1128/jvi.77.1.460-469.2003
Role of protein kinase C betaII in influenza virus entry via late endosomes
Abstract
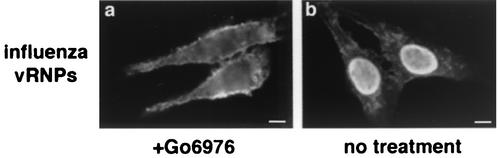
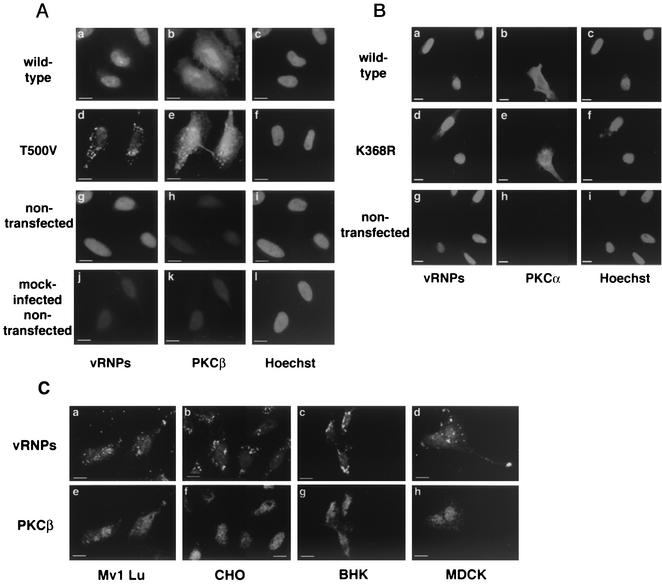
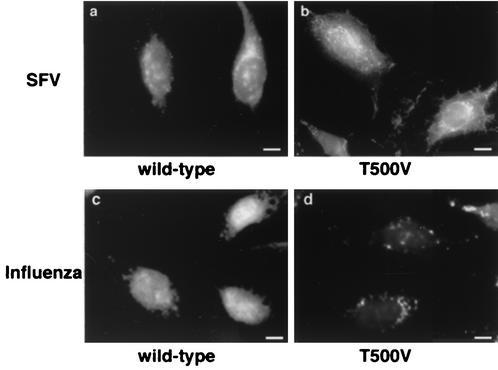
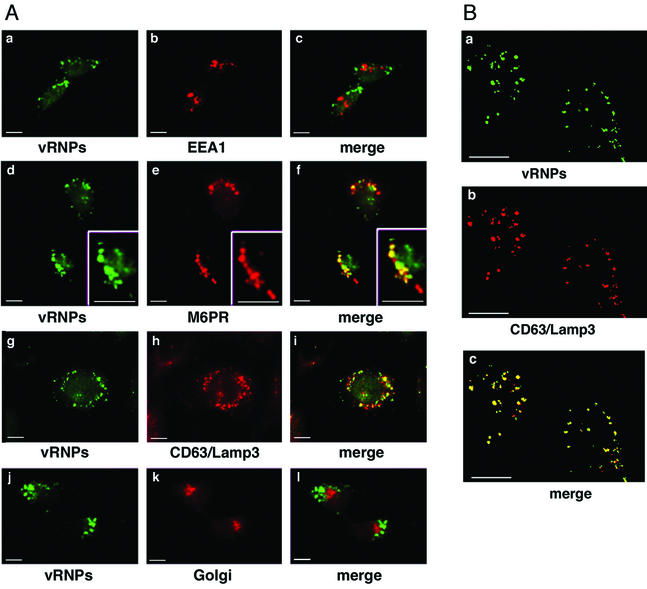
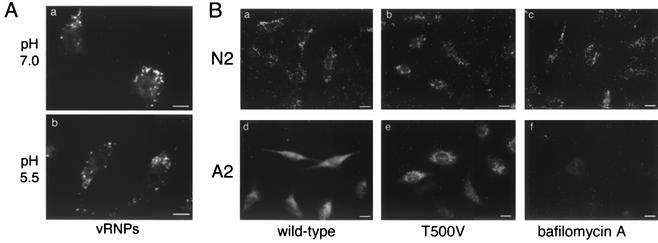
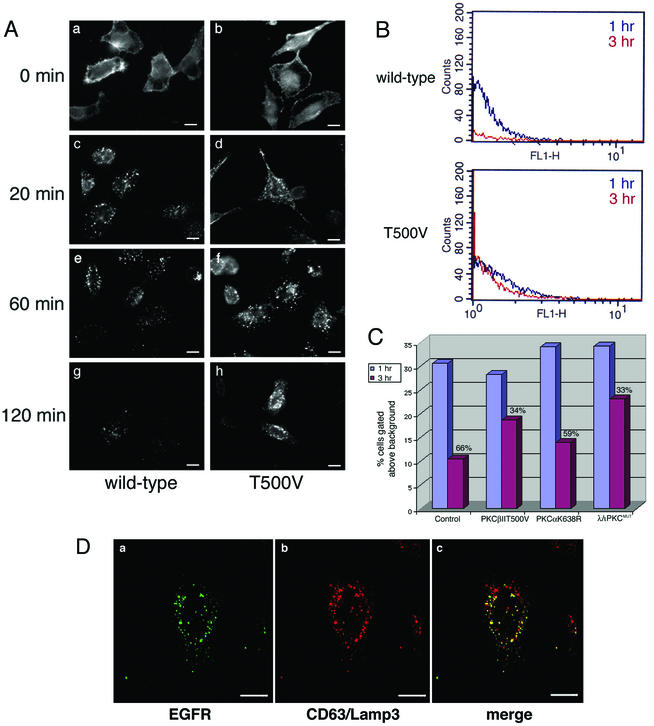
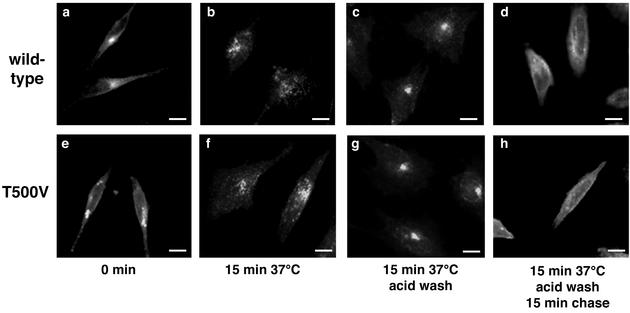
Many viruses take advantage of receptor-mediated endocytosis in order to enter target cells. We have utilized influenza virus and Semliki Forest virus (SFV) to define a role for protein kinase C betaII (PKCbetaII) in endocytic trafficking. We show that specific PKC inhibitors prevent influenza virus infection, suggesting a role for classical isoforms of PKC. We also examined virus entry in cells overexpressing dominant-negative forms of PKCalpha and -beta. Cells expressing a phosphorylation-deficient form of PKCbetaII (T500V), but not an equivalent mutant form of PKCalpha, inhibited successful influenza virus entry-with the virus accumulating in late endosomes. SFV, however, believed to enter cells from the early endosome, was unaffected by PKCbetaII T500V expression. We also examined the trafficking of two cellular ligands, transferrin and epidermal growth factor (EGF). PKCbetaII T500V expression specifically blocked EGF receptor trafficking and degradation, without affecting transferrin receptor recycling. As with influenza virus, in PKCbetaII kinase-dead cells, EGF receptor was trapped in a late endosome compartment. Our findings suggest that PKCbetaII is an important regulator of a late endosomal sorting event needed for influenza virus entry and infection.
Figures
Similar articles
-
Role of TSPAN9 in Alphavirus Entry and Early Endosomes.J Virol. 2016 Apr 14;90(9):4289-97. doi: 10.1128/JVI.00018-16. Print 2016 May. J Virol. 2016. PMID: 26865714 Free PMC article.
-
Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis.J Virol. 2019 May 29;93(12):e00017-19. doi: 10.1128/JVI.00017-19. Print 2019 Jun 15. J Virol. 2019. PMID: 30918081 Free PMC article.
-
Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis.J Virol. 2002 Oct;76(20):10455-64. doi: 10.1128/jvi.76.20.10455-10464.2002. J Virol. 2002. PMID: 12239322 Free PMC article.
-
Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses.Traffic. 2003 May;4(5):333-43. doi: 10.1034/j.1600-0854.2003.00090.x. Traffic. 2003. PMID: 12713661
-
Trafficking of the epidermal growth factor receptor and transferrin in three hepatocytic endosomal fractions.J Biol Chem. 1991 Jan 25;266(3):1396-402. J Biol Chem. 1991. PMID: 1671034
Cited by
-
Role of protein kinase C delta in reactivation of Kaposi's sarcoma-associated herpesvirus.J Virol. 2004 Sep;78(18):10187-92. doi: 10.1128/JVI.78.18.10187-10192.2004. J Virol. 2004. PMID: 15331751 Free PMC article.
-
Response Modifiers: Tweaking the Immune Response Against Influenza A Virus.Front Immunol. 2019 Apr 12;10:809. doi: 10.3389/fimmu.2019.00809. eCollection 2019. Front Immunol. 2019. PMID: 31031778 Free PMC article. Review.
-
Internalization of swine vesicular disease virus into cultured cells: a comparative study with foot-and-mouth disease virus.J Virol. 2009 May;83(9):4216-26. doi: 10.1128/JVI.02436-08. Epub 2009 Feb 18. J Virol. 2009. PMID: 19225001 Free PMC article.
-
Applying horizontal gene transfer phenomena to enhance non-viral gene therapy.J Control Release. 2013 Nov 28;172(1):246-257. doi: 10.1016/j.jconrel.2013.08.025. Epub 2013 Aug 30. J Control Release. 2013. PMID: 23994344 Free PMC article. Review.
-
Phosphoproteomic analysis reveals Smad protein family activation following Rift Valley fever virus infection.PLoS One. 2018 Feb 6;13(2):e0191983. doi: 10.1371/journal.pone.0191983. eCollection 2018. PLoS One. 2018. PMID: 29408900 Free PMC article.
References
-
- Benmerah, A., M. Bayrou, N. Cerf-Bensussan, and A. Dautry-Varsat. 1999. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J. Cell Sci. 112:1303-1311. - PubMed
-
- Breton, A., and A. Descoteaux. 2000. Protein kinase C-alpha participates in FcγR-mediated phagocytosis in macrophages. Biochem. Biophys. Res. Commun. 276:472-476. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
