

Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome
- PMID: 12369018
- PMCID: PMC419999
- DOI: 10.1086/342975
Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome
Abstract
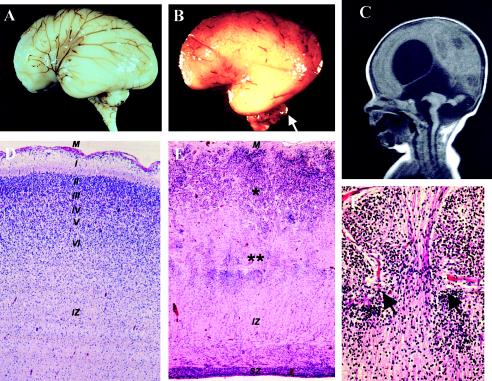
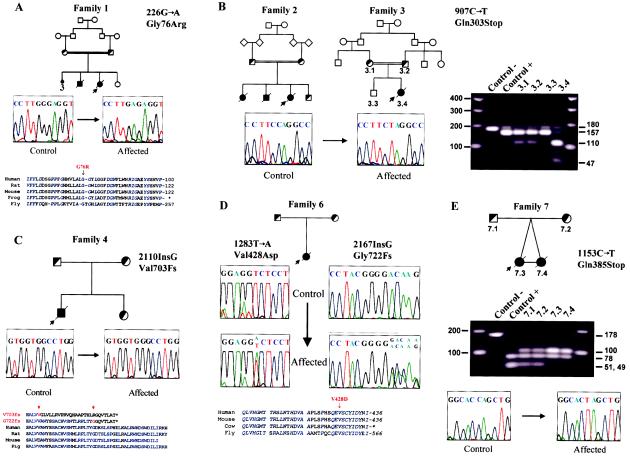
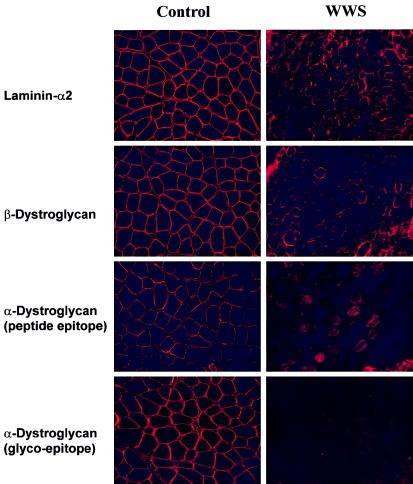
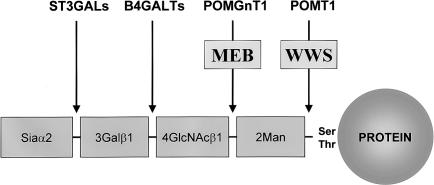
Walker-Warburg syndrome (WWS) is an autosomal recessive developmental disorder characterized by congenital muscular dystrophy and complex brain and eye abnormalities. A similar combination of symptoms is presented by two other human diseases, muscle-eye-brain disease (MEB) and Fukuyama congenital muscular dystrophy (FCMD). Although the genes underlying FCMD (Fukutin) and MEB (POMGnT1) have been cloned, loci for WWS have remained elusive. The protein products of POMGnT1 and Fukutin have both been implicated in protein glycosylation. To unravel the genetic basis of WWS, we first performed a genomewide linkage analysis in 10 consanguineous families with WWS. The results indicated the existence of at least three WWS loci. Subsequently, we adopted a candidate-gene approach in combination with homozygosity mapping in 15 consanguineous families with WWS. Candidate genes were selected on the basis of the role of the FCMD and MEB genes. Since POMGnT1 encodes an O-mannoside N-acetylglucosaminyltransferase, we analyzed the possible implication of O-mannosyl glycan synthesis in WWS. Analysis of the locus for O-mannosyltransferase 1 (POMT1) revealed homozygosity in 5 of 15 families. Sequencing of the POMT1 gene revealed mutations in 6 of the 30 unrelated patients with WWS. Of the five mutations identified, two are nonsense mutations, two are frameshift mutations, and one is a missense mutation. Immunohistochemical analysis of muscle from patients with POMT1 mutations corroborated the O-mannosylation defect, as judged by the absence of glycosylation of alpha-dystroglycan. The implication of O-mannosylation in MEB and WWS suggests new lines of study in understanding the molecular basis of neuronal migration.
Figures
Similar articles
-
POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome.J Med Genet. 2005 Dec;42(12):907-12. doi: 10.1136/jmg.2005.031963. Epub 2005 May 13. J Med Genet. 2005. PMID: 15894594 Free PMC article.
-
Expression and localization of fukutin, POMGnT1, and POMT1 in the central nervous system: consideration for functions of fukutin.Med Electron Microsc. 2004 Dec;37(4):200-7. doi: 10.1007/s00795-004-0260-5. Med Electron Microsc. 2004. PMID: 15614444 Review.
-
Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality.Proc Natl Acad Sci U S A. 2004 Sep 28;101(39):14126-31. doi: 10.1073/pnas.0405899101. Epub 2004 Sep 21. Proc Natl Acad Sci U S A. 2004. PMID: 15383666 Free PMC article.
-
Mutations of the POMT1 gene found in patients with Walker-Warburg syndrome lead to a defect of protein O-mannosylation.Biochem Biophys Res Commun. 2004 Dec 3;325(1):75-9. doi: 10.1016/j.bbrc.2004.10.001. Biochem Biophys Res Commun. 2004. PMID: 15522202
-
Fukuyama-type congenital muscular dystrophy (FCMD) and alpha-dystroglycanopathy.Congenit Anom (Kyoto). 2003 Jun;43(2):97-104. doi: 10.1111/j.1741-4520.2003.tb01033.x. Congenit Anom (Kyoto). 2003. PMID: 12893968 Review.
Cited by
-
Post-translational Modification in Muscular Dystrophies.Adv Exp Med Biol. 2022;1382:71-84. doi: 10.1007/978-3-031-05460-0_5. Adv Exp Med Biol. 2022. PMID: 36029404
-
Motor outcome measures in patients with FKRP mutations: A longitudinal follow-up.Neurology. 2020 Oct 13;95(15):e2131-e2139. doi: 10.1212/WNL.0000000000010604. Epub 2020 Aug 6. Neurology. 2020. PMID: 32764098 Free PMC article.
-
Fukutin is prerequisite to ameliorate muscular dystrophic phenotype by myofiber-selective LARGE expression.Sci Rep. 2015 Feb 9;5:8316. doi: 10.1038/srep08316. Sci Rep. 2015. PMID: 25661440 Free PMC article.
-
Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy.Eur J Hum Genet. 2012 Dec;20(12):1234-9. doi: 10.1038/ejhg.2012.71. Epub 2012 May 2. Eur J Hum Genet. 2012. PMID: 22549409 Free PMC article.
-
The role of pericytic laminin in blood brain barrier integrity maintenance.Sci Rep. 2016 Nov 3;6:36450. doi: 10.1038/srep36450. Sci Rep. 2016. PMID: 27808256 Free PMC article.
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for POMT2 [accession number AF105020])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for WWS [MIM 236670], MEB [MIM 253280], and FCMD [MIM 253800])
References
-
- Amado M, Almeida R, Schwientek T, Clausen H (1999) Identification and characterization of large galactosyltransferase gene families: galactosyltransferases for all functions. Biochim Biophys Acta 1473:35–53 - PubMed
-
- Anton ES, Kreidberg JA, Rakic P (1999) Distinct functions of α3 and αV integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron 22:277–289 - PubMed
-
- Aravind L, Koonin EV (1999) The fukutin protein family—predicted enzymes modifying cell-surface molecules. Curr Biol 9:R836–R837 - PubMed
-
- Barresi R, Confalonieri V, Lanfossi M, Di Blasi C, Torchiana E, Mantegazza R, Jarre L, Nardocci N, Boffi P, Tezzon F, Pini A, Cornelio F, Mora M, Morandi L (1997) Concomitant deficiency of β- and γ-sarcoglycans in 20 α-sarcoglycan (adhalin)-deficient patients: immunohistochemical analysis and clinical aspects. Acta Neuropathol (Berl) 94:28–35 - PubMed
-
- Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB, Mercuri E, Voit T, Sewry CA, Guicheney P, Muntoni F (2001a) Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am J Hum Genet 69:1198–1209 - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
