

Comparative genomics and evolution of proteins involved in RNA metabolism
- PMID: 11917006
- PMCID: PMC101826
- DOI: 10.1093/nar/30.7.1427
Comparative genomics and evolution of proteins involved in RNA metabolism
Abstract
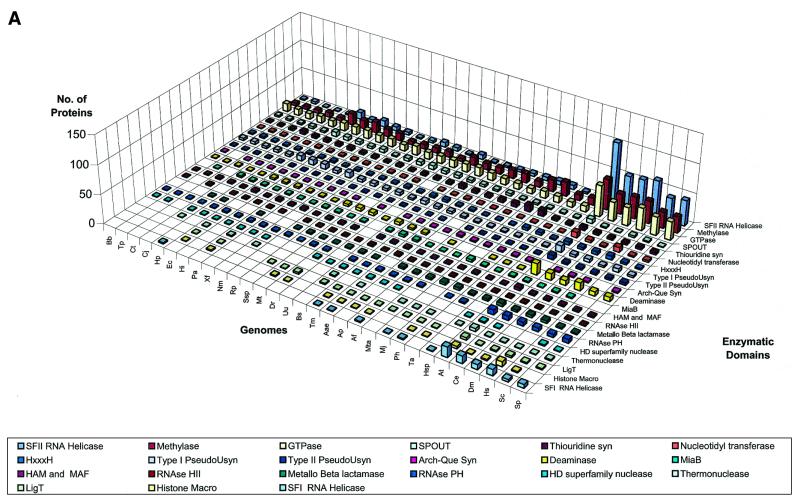
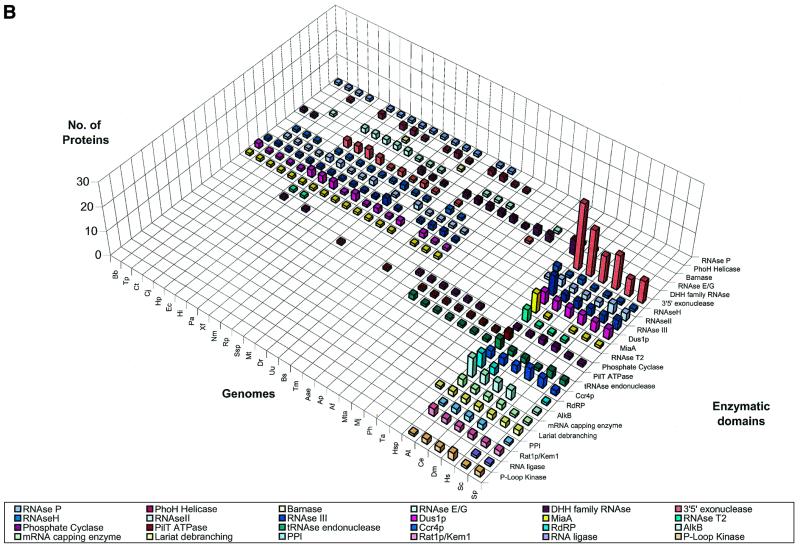
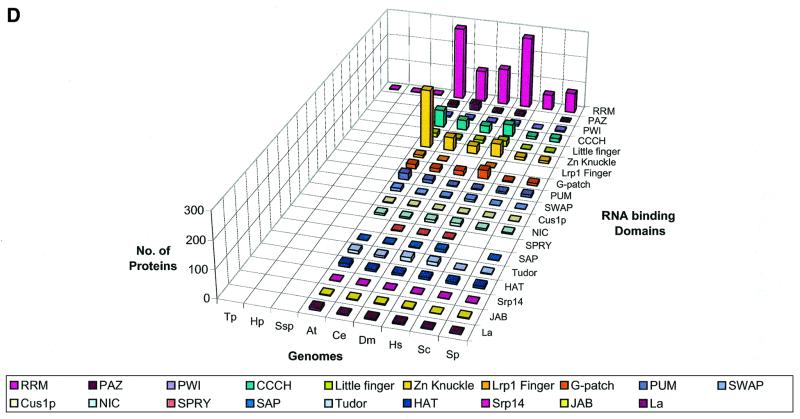
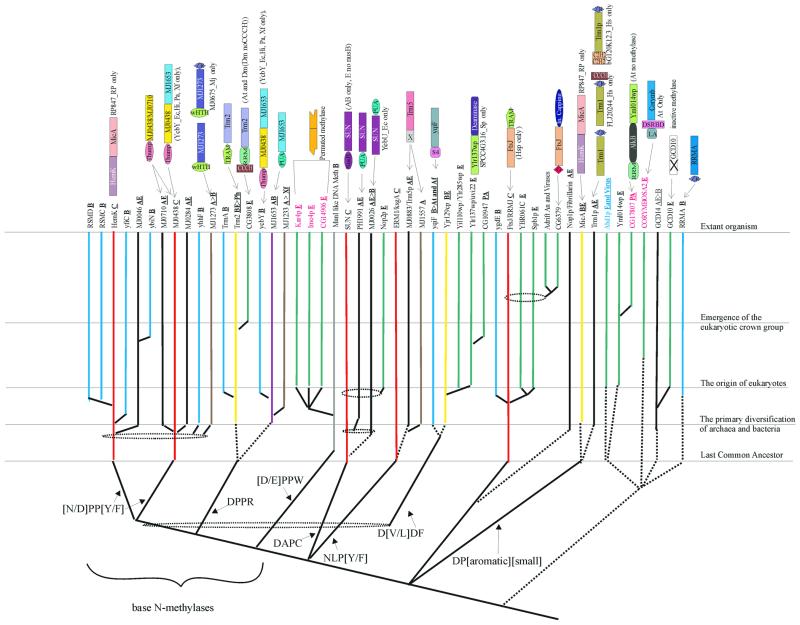
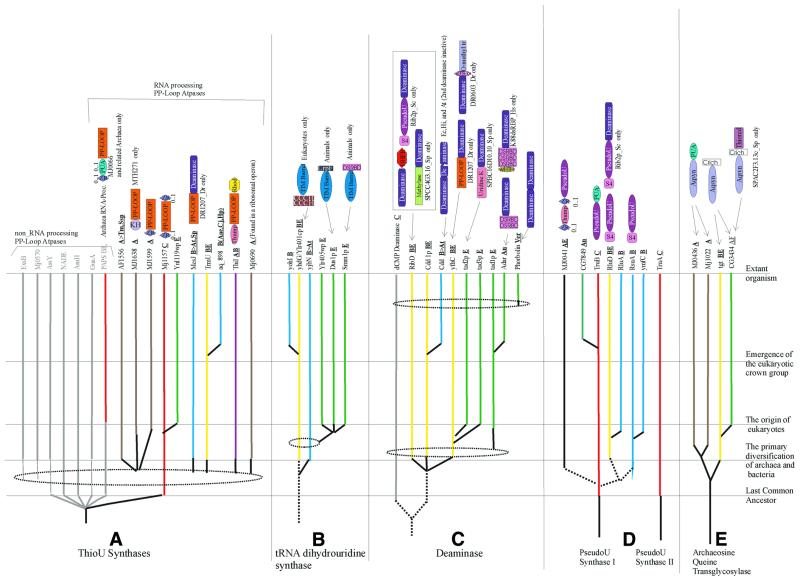
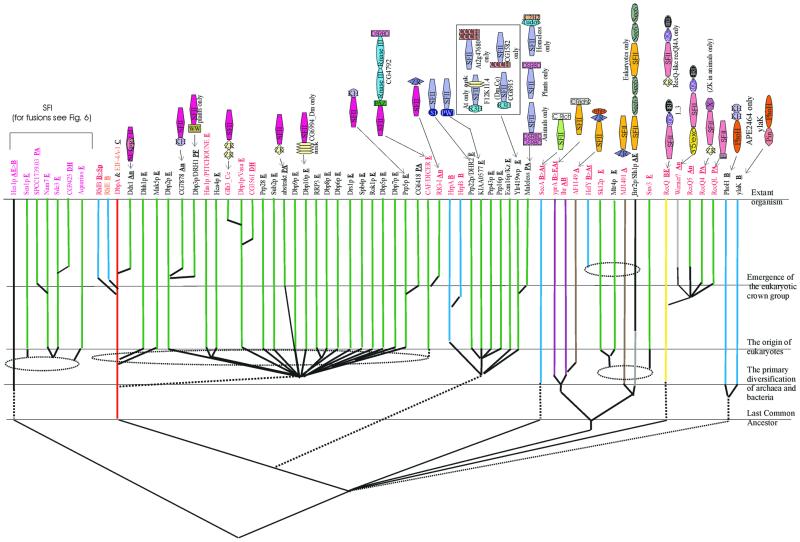
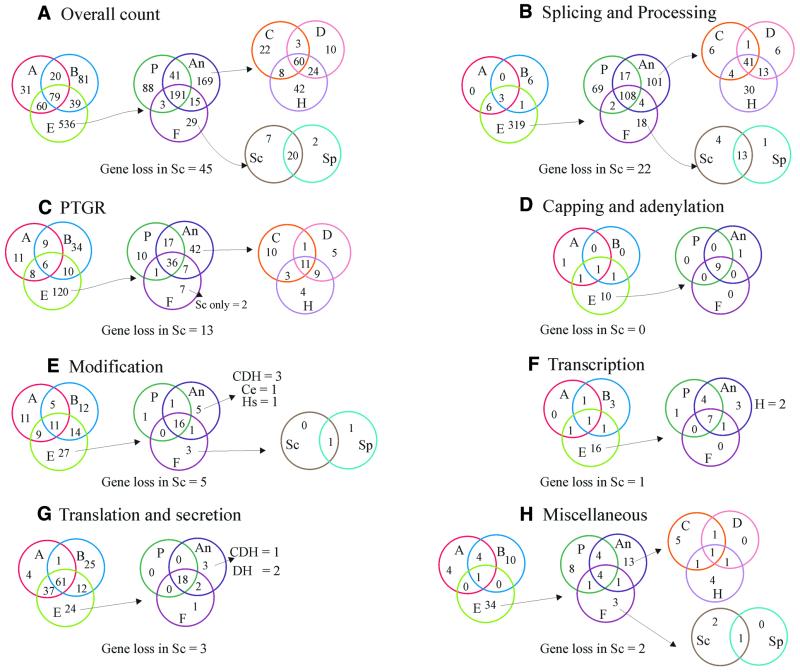
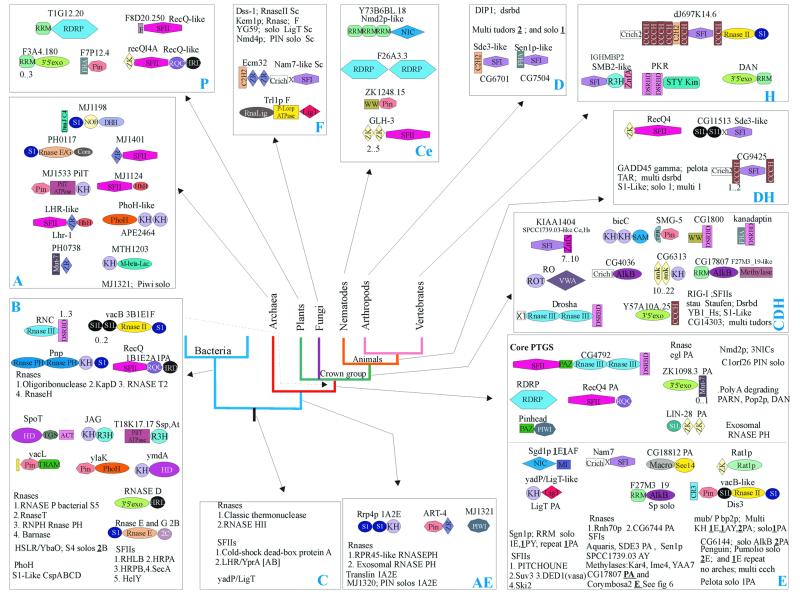
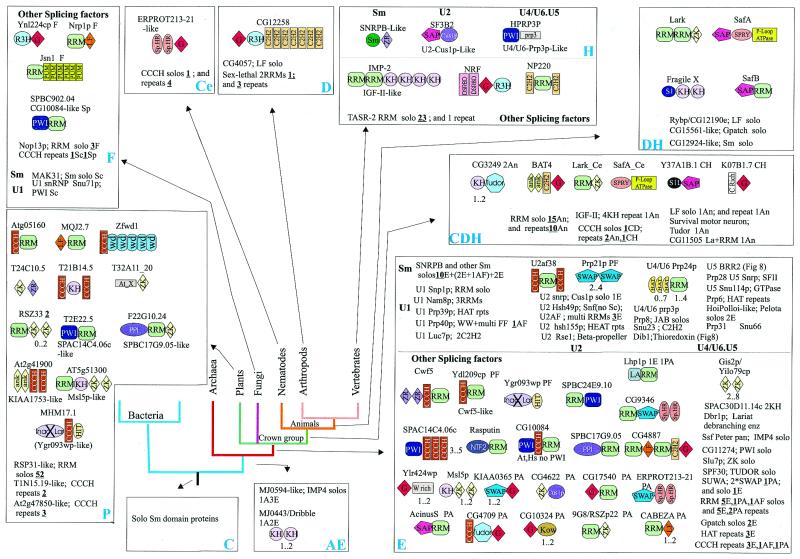
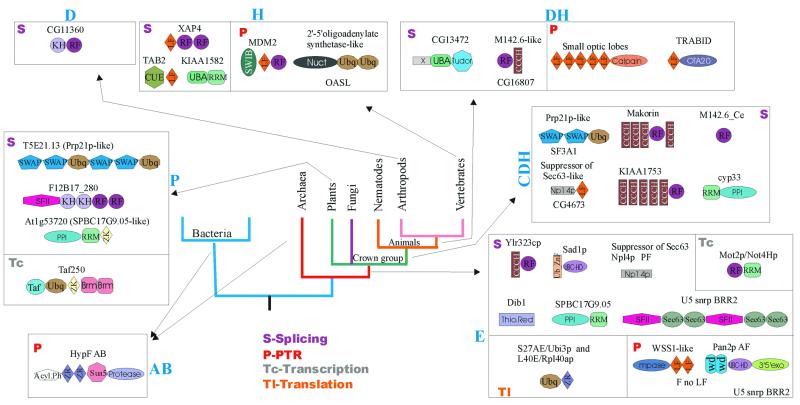
RNA metabolism, broadly defined as the compendium of all processes that involve RNA, including transcription, processing and modification of transcripts, translation, RNA degradation and its regulation, is the central and most evolutionarily conserved part of cell physiology. A comprehensive, genome-wide census of all enzymatic and non-enzymatic protein domains involved in RNA metabolism was conducted by using sequence profile analysis and structural comparisons. Proteins related to RNA metabolism comprise from 3 to 11% of the complete protein repertoire in bacteria, archaea and eukaryotes, with the greatest fraction seen in parasitic bacteria with small genomes. Approximately one-half of protein domains involved in RNA metabolism are present in most, if not all, species from all three primary kingdoms and are traceable to the last universal common ancestor (LUCA). The principal features of LUCA's RNA metabolism system were reconstructed by parsimony-based evolutionary analysis of all relevant groups of orthologous proteins. This reconstruction shows that LUCA possessed not only the basal translation system, but also the principal forms of RNA modification, such as methylation, pseudouridylation and thiouridylation, as well as simple mechanisms for polyadenylation and RNA degradation. Some of these ancient domains form paralogous groups whose evolution can be traced back in time beyond LUCA, towards low-specificity proteins, which probably functioned as cofactors for ribozymes within the RNA world framework. The main lineage-specific innovations of RNA metabolism systems were identified. The most notable phase of innovation in RNA metabolism coincides with the advent of eukaryotes and was brought about by the merge of the archaeal and bacterial systems via mitochondrial endosymbiosis, but also involved emergence of several new, eukaryote-specific RNA-binding domains. Subsequent, vast expansions of these domains mark the origin of alternative splicing in animals and probably in plants. In addition to the reconstruction of the evolutionary history of RNA metabolism, this analysis produced numerous functional predictions, e.g. of previously undetected enzymes of RNA modification.
Figures

Similar articles
-
The many faces of the helix-turn-helix domain: transcription regulation and beyond.FEMS Microbiol Rev. 2005 Apr;29(2):231-62. doi: 10.1016/j.femsre.2004.12.008. FEMS Microbiol Rev. 2005. PMID: 15808743 Review.
-
CoCoNuTs are a diverse subclass of Type IV restriction systems predicted to target RNA.Elife. 2024 May 13;13:RP94800. doi: 10.7554/eLife.94800. Elife. 2024. PMID: 38739430 Free PMC article.
-
Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases.BMC Struct Biol. 2003 Jan 28;3:1. doi: 10.1186/1472-6807-3-1. Epub 2003 Jan 28. BMC Struct Biol. 2003. PMID: 12553882 Free PMC article.
-
Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes.BMC Evol Biol. 2003 Jan 6;3:2. doi: 10.1186/1471-2148-3-2. Epub 2003 Jan 6. BMC Evol Biol. 2003. PMID: 12515582 Free PMC article.
-
The last universal common ancestor: emergence, constitution and genetic legacy of an elusive forerunner.Biol Direct. 2008 Jul 9;3:29. doi: 10.1186/1745-6150-3-29. Biol Direct. 2008. PMID: 18613974 Free PMC article. Review.
Cited by
-
Roquin binds microRNA-146a and Argonaute2 to regulate microRNA homeostasis.Nat Commun. 2015 Feb 20;6:6253. doi: 10.1038/ncomms7253. Nat Commun. 2015. PMID: 25697406 Free PMC article.
-
On the origin of the translation system and the genetic code in the RNA world by means of natural selection, exaptation, and subfunctionalization.Biol Direct. 2007 May 31;2:14. doi: 10.1186/1745-6150-2-14. Biol Direct. 2007. PMID: 17540026 Free PMC article.
-
A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes.Genome Biol. 2004;5(2):R7. doi: 10.1186/gb-2004-5-2-r7. Epub 2004 Jan 15. Genome Biol. 2004. PMID: 14759257 Free PMC article.
-
The DEAD-Box RNA Helicases of Bacillus subtilis as a Model to Evaluate Genetic Compensation Among Duplicate Genes.Front Microbiol. 2018 Sep 25;9:2261. doi: 10.3389/fmicb.2018.02261. eCollection 2018. Front Microbiol. 2018. PMID: 30337909 Free PMC article.
-
The last common ancestor: what's in a name?Orig Life Evol Biosph. 2005 Dec;35(6):537-54. doi: 10.1007/s11084-005-5760-3. Orig Life Evol Biosph. 2005. PMID: 16254691
References
-
- Alberts B., Bray,D., Lewis,J., Raff,M., Roberts,K. and Watson,J.D. (1999) Molecular Biology of the Cell. Garland Publishing, New York, NY.
-
- Crick F.H.C. (1958) Symp. Soc. Exp. Biol. XII, 139–163.
-
- Reinhart B.J., Slack,F.J., Basson,M., Pasquinelli,A.E., Bettinger,J.C., Rougvie,A.E., Horvitz,H.R. and Ruvkun,G. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature, 403, 901–906. - PubMed
-
- Franke A. and Baker,B.S. (1999) The rox1 and rox2 RNAs are essential components of the compensasome, which mediates dosage compensation in Drosophila. Mol. Cell, 4, 117–122. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
