

APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death
- PMID: 11591730
- PMCID: PMC2198828
- DOI: 10.1083/jcb.200105137
APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death
Abstract
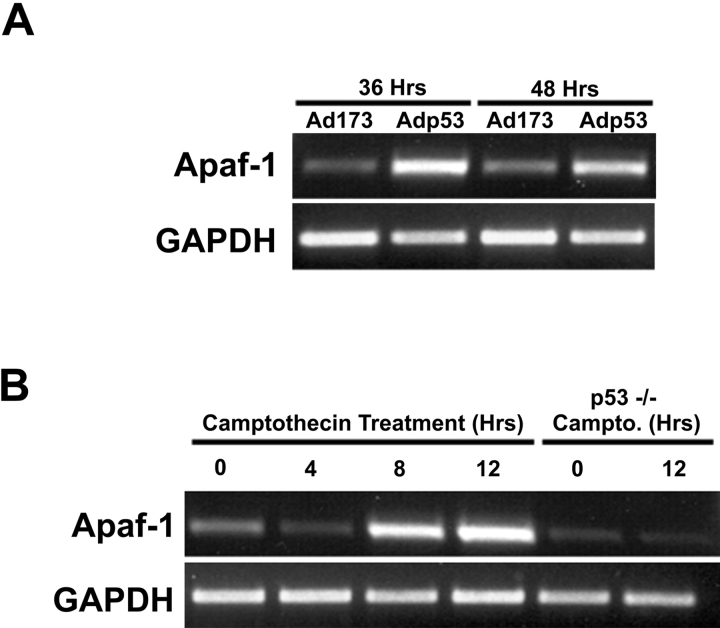
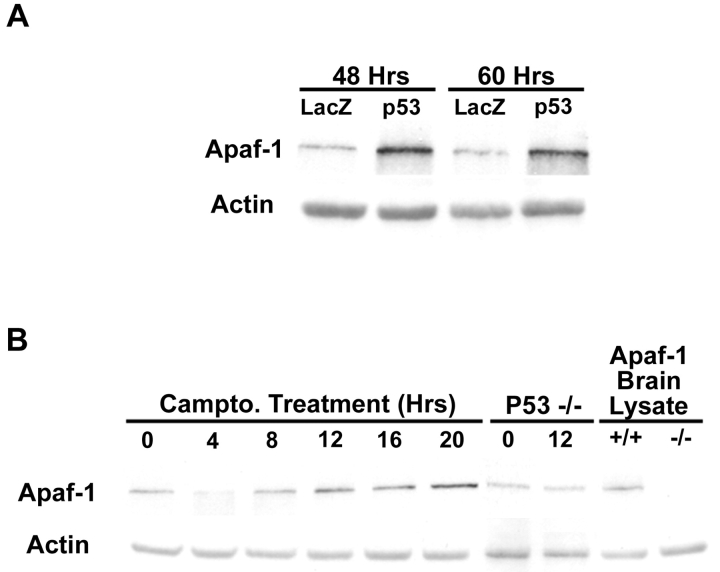
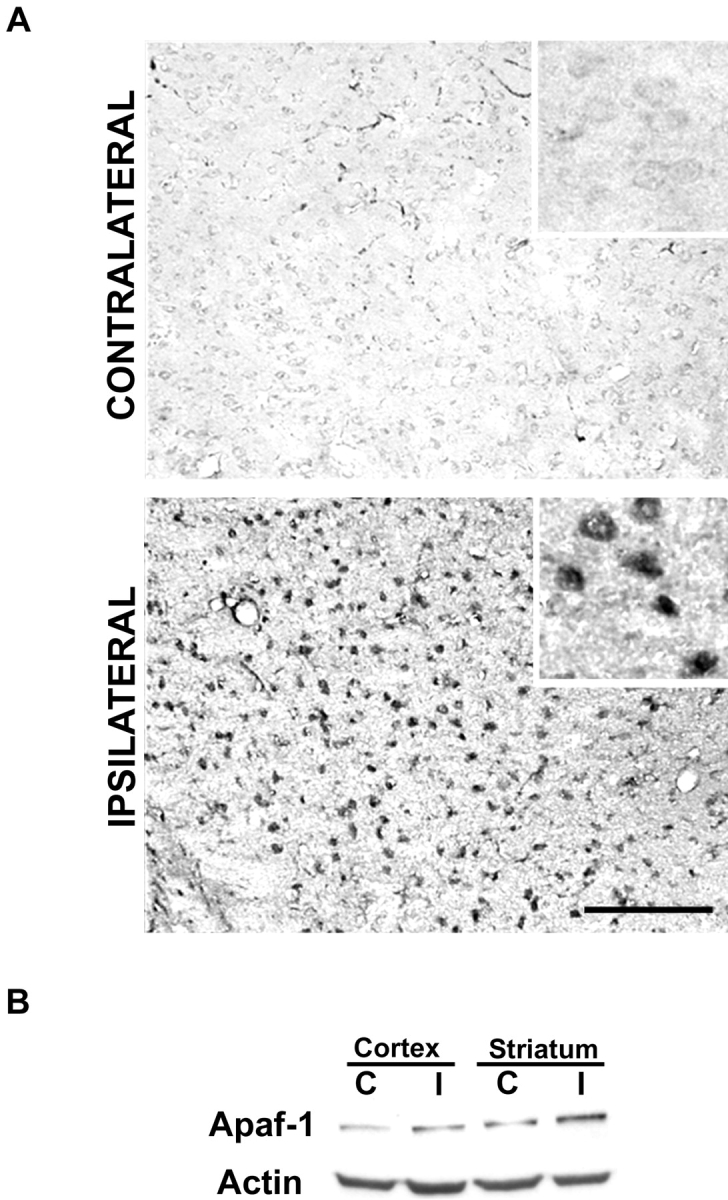
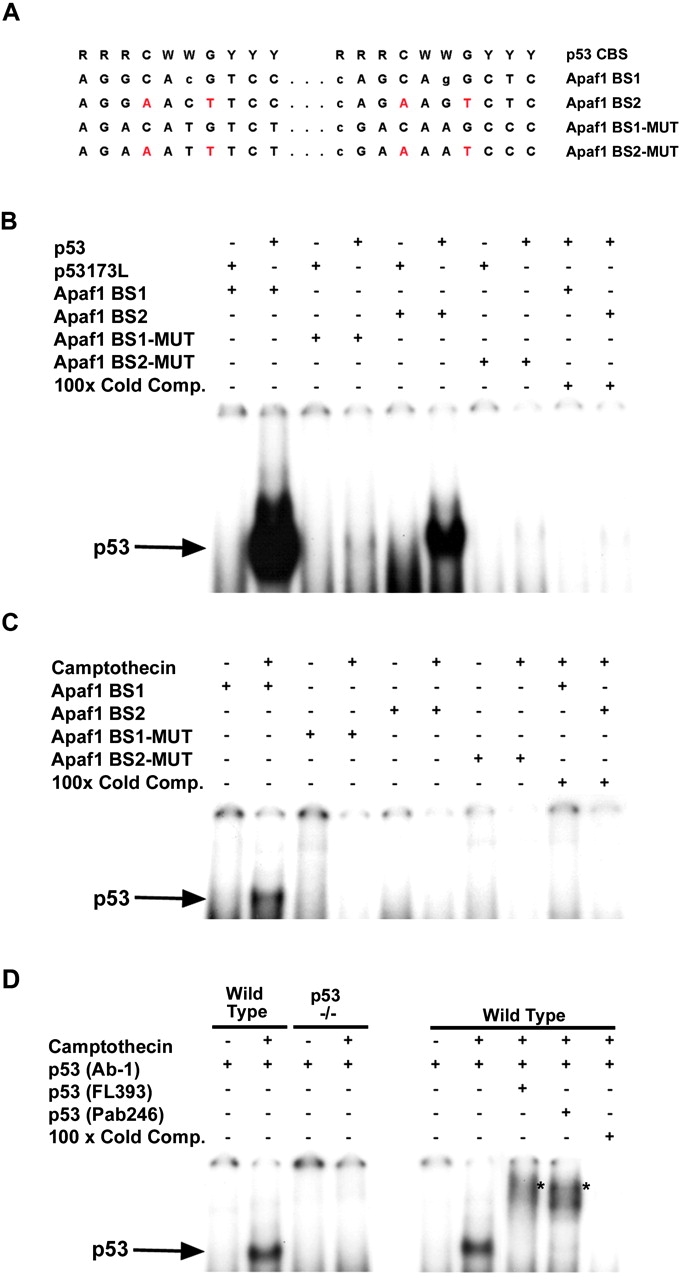
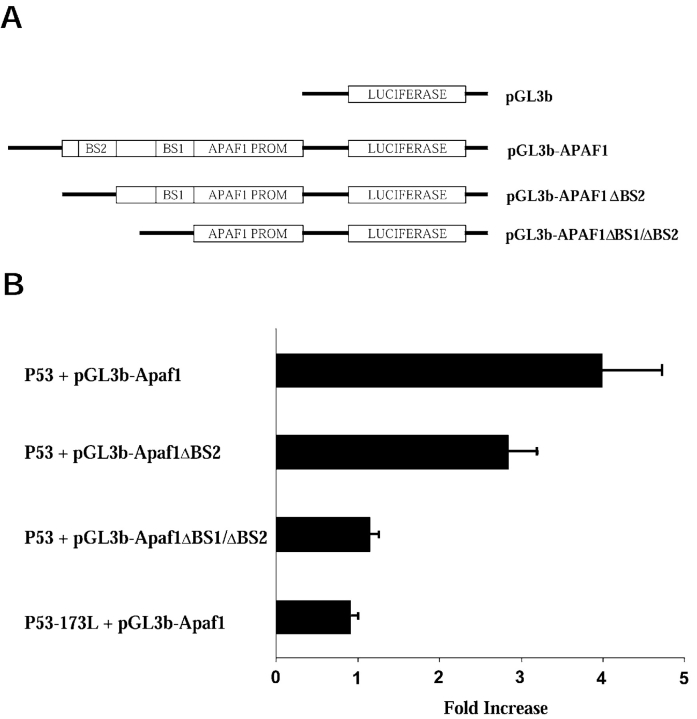
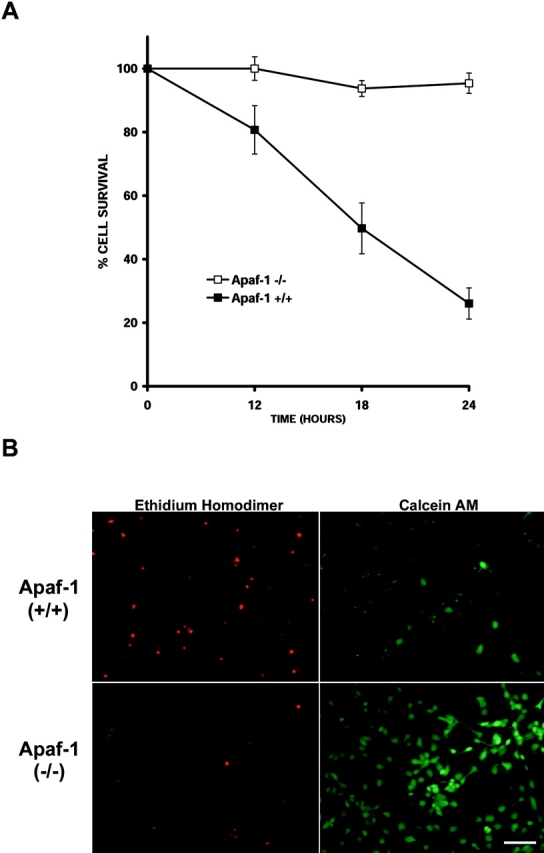
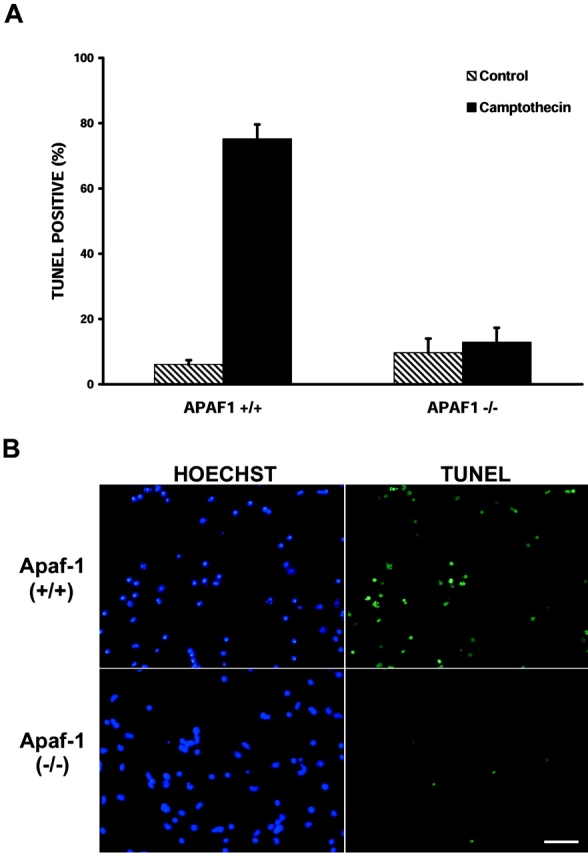
p53 is a transcriptional activator which has been implicated as a key regulator of neuronal cell death after acute injury. We have shown previously that p53-mediated neuronal cell death involves a Bax-dependent activation of caspase 3; however, the transcriptional targets involved in the regulation of this process have not been identified. In the present study, we demonstrate that p53 directly upregulates Apaf1 transcription as a critical step in the induction of neuronal cell death. Using DNA microarray analysis of total RNA isolated from neurons undergoing p53-induced apoptosis a 5-6-fold upregulation of Apaf1 mRNA was detected. Induction of neuronal cell death by camptothecin, a DNA-damaging agent that functions through a p53-dependent mechanism, resulted in increased Apaf1 mRNA in p53-positive, but not p53-deficient neurons. In both in vitro and in vivo neuronal cell death processes of p53-induced cell death, Apaf1 protein levels were increased. We addressed whether p53 directly regulates Apaf1 transcription via the two p53 consensus binding sites in the Apaf1 promoter. Electrophoretic mobility shift assays demonstrated p53-DNA binding activity at both p53 consensus binding sequences in extracts obtained from neurons undergoing p53-induced cell death, but not in healthy control cultures or when p53 or the p53 binding sites were inactivated by mutation. In transient transfections in a neuronal cell line with p53 and Apaf1 promoter-luciferase constructs, p53 directly activated the Apaf1 promoter via both p53 sites. The importance of Apaf1 as a p53 target gene in neuronal cell death was evaluated by examining p53-induced apoptotic pathways in primary cultures of Apaf1-deficient neurons. Neurons treated with camptothecin were significantly protected in the absence of Apaf1 relative to those derived from wild-type littermates. Together, these results demonstrate that Apaf1 is a key transcriptional target for p53 that plays a pivotal role in the regulation of apoptosis after neuronal injury.
Figures
Similar articles
-
Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death.J Cell Biol. 2002 Aug 5;158(3):507-17. doi: 10.1083/jcb.200202130. Epub 2002 Jul 29. J Cell Biol. 2002. PMID: 12147675 Free PMC article.
-
The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1.J Biol Chem. 2004 Jul 2;279(27):28706-14. doi: 10.1074/jbc.M400376200. Epub 2004 Apr 22. J Biol Chem. 2004. PMID: 15105421
-
Transcriptional activation of APAF1 by KAISO (ZBTB33) and p53 is attenuated by RelA/p65.Biochim Biophys Acta. 2015 Sep;1849(9):1170-8. doi: 10.1016/j.bbagrm.2015.07.008. Epub 2015 Jul 14. Biochim Biophys Acta. 2015. PMID: 26183023
-
The regulation of APAF1 expression during development and tumourigenesis.Apoptosis. 2002 Apr;7(2):167-71. doi: 10.1023/a:1014370616864. Apoptosis. 2002. PMID: 11865201 Review.
-
Apaf1 in developmental apoptosis and cancer: how many ways to die?Cell Mol Life Sci. 2001 Oct;58(11):1688-97. doi: 10.1007/pl00000806. Cell Mol Life Sci. 2001. PMID: 11706994 Free PMC article. Review.
Cited by
-
Versatile functions of p53 protein in multicellular organisms.Biochemistry (Mosc). 2007 Dec;72(13):1399-421. doi: 10.1134/s0006297907130019. Biochemistry (Mosc). 2007. PMID: 18282133 Free PMC article. Review.
-
Nuclear factor-(kappa)B modulates the p53 response in neurons exposed to DNA damage.J Neurosci. 2004 Mar 24;24(12):2963-73. doi: 10.1523/JNEUROSCI.0155-04.2004. J Neurosci. 2004. PMID: 15044535 Free PMC article.
-
Borna disease virus phosphoprotein represses p53-mediated transcriptional activity by interference with HMGB1.J Virol. 2003 Nov;77(22):12243-51. doi: 10.1128/jvi.77.22.12243-12251.2003. J Virol. 2003. PMID: 14581561 Free PMC article.
-
Tumor suppressor p53 modulates activity-dependent synapse strengthening, autism-like behavior and hippocampus-dependent learning.Mol Psychiatry. 2023 Sep;28(9):3782-3794. doi: 10.1038/s41380-023-02268-9. Epub 2023 Sep 28. Mol Psychiatry. 2023. PMID: 37759036 Free PMC article.
-
Synaptic activity-mediated suppression of p53 and induction of nuclear calcium-regulated neuroprotective genes promote survival through inhibition of mitochondrial permeability transition.J Neurosci. 2009 Apr 8;29(14):4420-9. doi: 10.1523/JNEUROSCI.0802-09.2009. J Neurosci. 2009. PMID: 19357269 Free PMC article.
References
-
- Banasiak, K.J., and G.G. Haddad. 1998. Hypoxia-induced apoptosis: effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res. 797:295–304. - PubMed
-
- Bates, S., A.C. Phillips, P.A. Clark, F. Stott, G. Peters, R.L. Ludwig, and K.H. Vousden. 1998. p14ARF links the tumour suppressors RB and p53. Nature. 395:124–125. - PubMed
-
- Cecconi, F., G. Alvarez-Bolado, B.I. Meyer, K.A. Roth, and P. Gruss. 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 94:727–737. - PubMed
-
- Cheng, Y., M. Deshmukh, A. D' Costa, J.A. Demaro, J.M. Gidday, A. Shah, Y. Sun, M.F. Jacquin, E.M. Johnson, and D.M. Holtzman. 1998. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J. Clin. Invest. 101:1992–1999. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
