

Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy
- PMID: 11443545
- PMCID: PMC1235303
- DOI: 10.1086/321295
Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy
Abstract
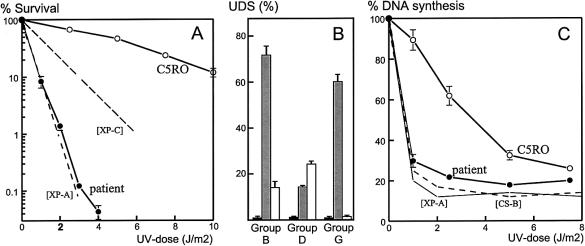
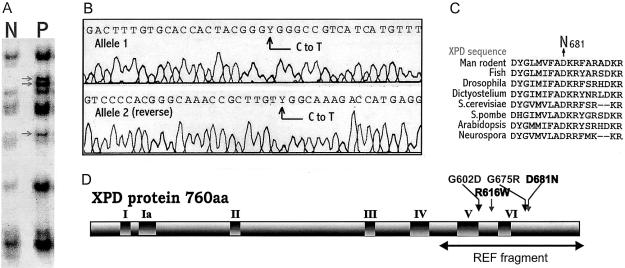
Cerebro-oculo-facio-skeletal (COFS) syndrome is a recessively inherited rapidly progressive neurologic disorder leading to brain atrophy, with calcifications, cataracts, microcornea, optic atrophy, progressive joint contractures, and growth failure. Cockayne syndrome (CS) is a recessively inherited neurodegenerative disorder characterized by low to normal birth weight, growth failure, brain dysmyelination with calcium deposits, cutaneous photosensitivity, pigmentary retinopathy and/or cataracts, and sensorineural hearing loss. Cultured CS cells are hypersensitive to UV radiation, because of impaired nucleotide-excision repair (NER) of UV-induced damage in actively transcribed DNA, whereas global genome NER is unaffected. The abnormalities in CS are caused by mutated CSA or CSB genes. Another class of patients with CS symptoms have mutations in the XPB, XPD, or XPG genes, which result in UV hypersensitivity as well as defective global NER; such patients may concurrently have clinical features of another NER syndrome, xeroderma pigmentosum (XP). Clinically observed similarities between COFS syndrome and CS have been followed by discoveries of cases of COFS syndrome that are associated with mutations in the XPG and CSB genes. Here we report the first involvement of the XPD gene in a new case of UV-sensitive COFS syndrome, with heterozygous substitutions-a R616W null mutation (previously seen in patients in XP complementation group D) and a unique D681N mutation-demonstrating that a third gene can be involved in COFS syndrome. We propose that COFS syndrome be included within the already known spectrum of NER disorders: XP, CS, and trichothiodystrophy. We predict that future patients with COFS syndrome will be found to have mutations in the CSA or XPB genes, and we document successful use of DNA repair for prenatal diagnosis in triplet and singleton pregnancies at risk for COFS syndrome. This result strongly underlines the need for screening of patients with COFS syndrome, for either UV sensitivity or DNA-repair abnormalities.
Figures

Similar articles
-
Manitoba aboriginal kindred with original cerebro-oculo- facio-skeletal syndrome has a mutation in the Cockayne syndrome group B (CSB) gene.Am J Hum Genet. 2000 Apr;66(4):1221-8. doi: 10.1086/302867. Epub 2000 Mar 15. Am J Hum Genet. 2000. PMID: 10739753 Free PMC article.
-
Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome.Hum Mutat. 2006 Nov;27(11):1092-103. doi: 10.1002/humu.20392. Hum Mutat. 2006. PMID: 16947863
-
Cerebro-oculo-facio-skeletal syndrome.Adv Exp Med Biol. 2010;685:210-4. doi: 10.1007/978-1-4419-6448-9_19. Adv Exp Med Biol. 2010. PMID: 20687508 Review.
-
Functional and molecular genetic analyses of nine newly identified XPD-deficient patients reveal a novel mutation resulting in TTD as well as in XP/CS complex phenotypes.Exp Dermatol. 2013 Jul;22(7):486-9. doi: 10.1111/exd.12166. Exp Dermatol. 2013. PMID: 23800062
-
Heterogeneity and overlaps in nucleotide excision repair disorders.Clin Genet. 2020 Jan;97(1):12-24. doi: 10.1111/cge.13545. Epub 2019 Apr 22. Clin Genet. 2020. PMID: 30919937 Review.
Cited by
-
First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure.Am J Hum Genet. 2007 Mar;80(3):457-66. doi: 10.1086/512486. Epub 2007 Jan 29. Am J Hum Genet. 2007. PMID: 17273966 Free PMC article.
-
Uncommon nucleotide excision repair phenotypes revealed by targeted high-throughput sequencing.Orphanet J Rare Dis. 2016 Mar 22;11:26. doi: 10.1186/s13023-016-0408-0. Orphanet J Rare Dis. 2016. PMID: 27004399 Free PMC article.
-
Phenotype-specific adverse effects of XPD mutations on human prenatal development implicate impairment of TFIIH-mediated functions in placenta.Eur J Hum Genet. 2012 Jun;20(6):626-31. doi: 10.1038/ejhg.2011.249. Epub 2012 Jan 11. Eur J Hum Genet. 2012. PMID: 22234153 Free PMC article.
-
Disease-causing missense mutations in human DNA helicase disorders.Mutat Res. 2013 Apr-Jun;752(2):138-152. doi: 10.1016/j.mrrev.2012.12.004. Epub 2012 Dec 28. Mutat Res. 2013. PMID: 23276657 Free PMC article. Review.
-
Transcription-associated breaks in xeroderma pigmentosum group D cells from patients with combined features of xeroderma pigmentosum and Cockayne syndrome.Mol Cell Biol. 2005 Sep;25(18):8368-78. doi: 10.1128/MCB.25.18.8368-8378.2005. Mol Cell Biol. 2005. PMID: 16135823 Free PMC article.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for COFS syndrome [MIM 214150]; CKN1, CSA, and CS [MIM 216400–MIM 216411]; CAMFAK [MIM 212540]; NLS [MIM 256520]; Martsolf [MIM 212720]; CAHMR [MIM 211770]; MICRO [MIM 600118]; XPA, XP, XP1, XPAC, XPD, XPDC, and XP4 [MIM 278700–MIM 278810]; and TTD [MIM 601675])
References
-
- Del Bigio MR, Greenberg CR, Rorke LB, Schnur R, McDonald-McGinn DM, Zackai EH (1997) Neuropathological findings in eight children with cerebro-oculo-facio-skeletal (COFS) syndrome. J Neuropathol Exp Neurol 56:1147–1157 - PubMed
-
- Hennekam RCM, van de Meeberg AG, van Doorne JM, Dijkstra PF, Bijlsma JB (1988) Martsolf syndrome in a brother and sister: clinical features and pattern of inheritance. Eur J Pediatr 147:539–543 - PubMed
-
- Lehmann AR (2001) The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev 15:15–23 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
