

IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB
- PMID: 11390419
- PMCID: PMC209314
- DOI: 10.1172/JCI10530
IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB
Abstract
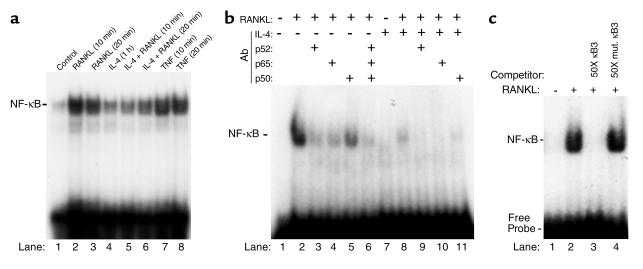
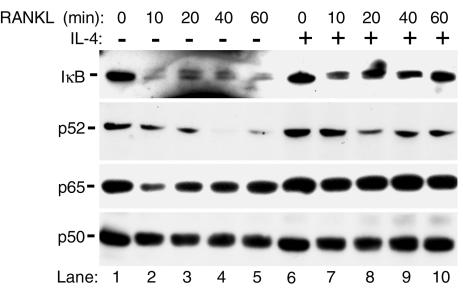
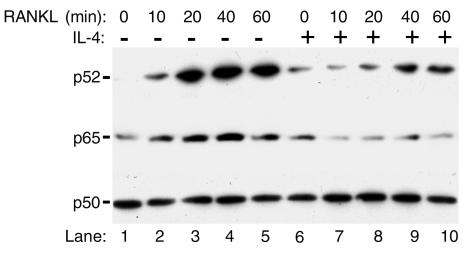
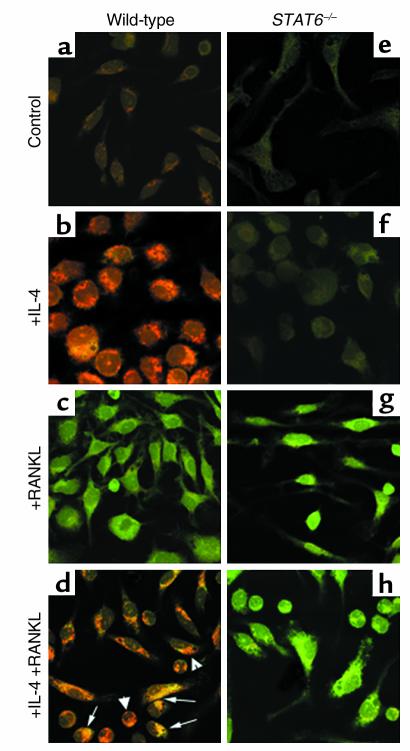
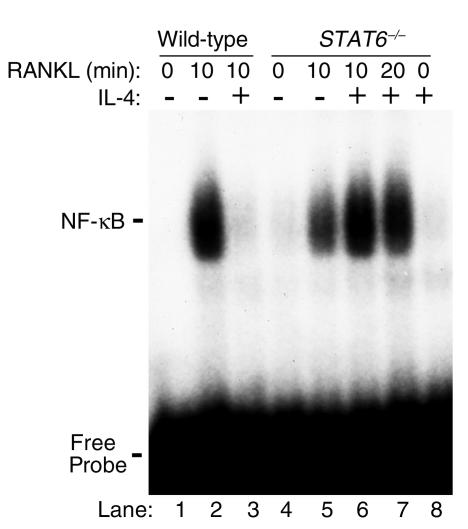
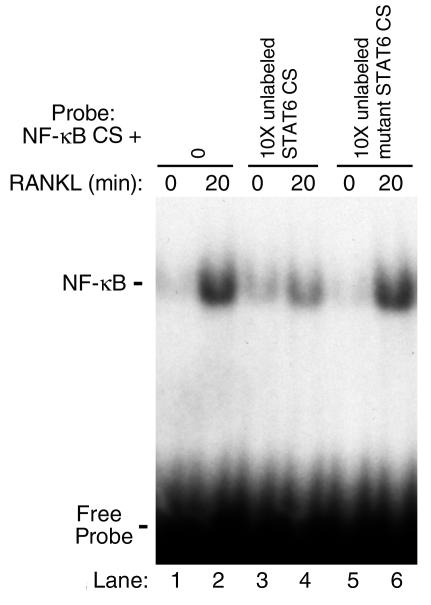
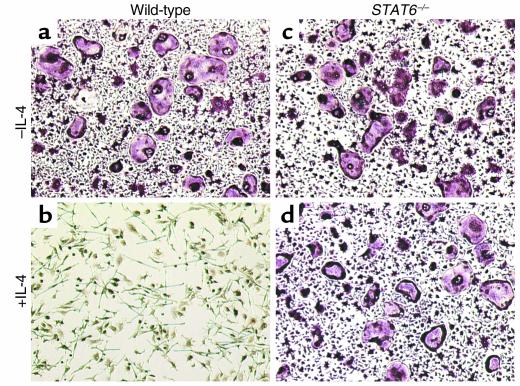
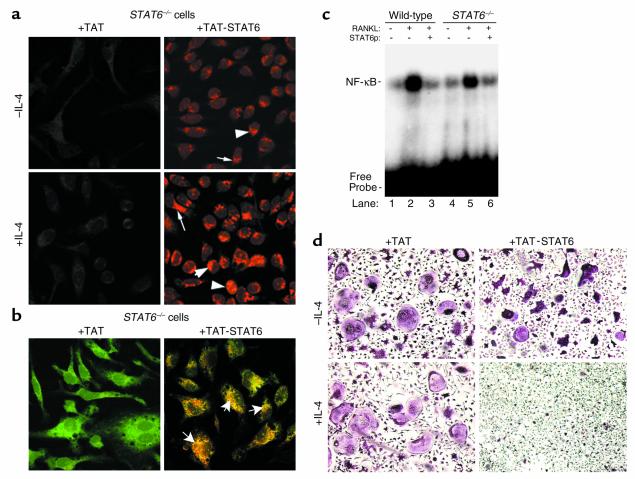
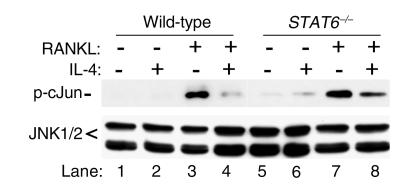
IL-4, an anti-inflammatory cytokine, inhibits osteoclast differentiation, but the basis of this effect has been unclear. Osteoclastogenesis requires activation of RANK, which exerts its biologic effect via activation of NF-kappaB. NF-kappaB activation is manifested by nuclear translocation and binding to DNA, events secondary to phosphorylation and dissociation of IkappaBalpha. It is shown here that IL-4 reduces NF-kappaB nuclear translocation by inhibiting IkappaB phosphorylation, thus markedly inhibiting NF-kappaB DNA binding activity and blocking osteoclastogenesis entirely. Residual translocation of NF-kappaB in the presence of IL-4, however, suggests that nuclear mechanisms must primarily account for inhibition of NF-kappaB DNA binding and blockade of osteoclastogenesis. To address this issue, this study examined whether IL-4-induced STAT6 transcription factor blocks NF-kappaB transactivation. The results show that excess unlabeled consensus sequence STAT6, but not its mutated form, inhibits NF-kappaB binding. Furthermore, exogenously added STAT6 protein inhibits NF-kappaB/DNA interaction. Further supporting a role for STAT6 in this process are the findings that IL-4 fails to block osteoclastogenesis in STAT6(-/-) mice but that this blockade can be restored with addition of exogenous STAT6. Thus, IL-4 obliterates osteoclast differentiation by antagonizing NF-kappaB activation in a STAT6-dependent manner.
Figures

Similar articles
-
Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways.J Biol Chem. 2001 Jan 5;276(1):563-8. doi: 10.1074/jbc.M008198200. J Biol Chem. 2001. PMID: 11032840
-
IL-3 acts directly on osteoclast precursors and irreversibly inhibits receptor activator of NF-kappa B ligand-induced osteoclast differentiation by diverting the cells to macrophage lineage.J Immunol. 2003 Jul 1;171(1):142-51. doi: 10.4049/jimmunol.171.1.142. J Immunol. 2003. PMID: 12816992
-
Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors.J Cell Biochem. 2003 Sep 1;90(1):59-67. doi: 10.1002/jcb.10613. J Cell Biochem. 2003. PMID: 12938156
-
Inhibition of inflammatory bone erosion by constitutively active STAT-6 through blockade of JNK and NF-kappaB activation.Arthritis Rheum. 2005 Sep;52(9):2719-29. doi: 10.1002/art.21286. Arthritis Rheum. 2005. PMID: 16142755
-
Current topics in pharmacological research on bone metabolism: osteoclast differentiation regulated by glycosphingolipids.J Pharmacol Sci. 2006 Mar;100(3):195-200. doi: 10.1254/jphs.fmj05004x3. Epub 2006 Mar 14. J Pharmacol Sci. 2006. PMID: 16538029 Review.
Cited by
-
The Autophagy in Osteoimmonology: Self-Eating, Maintenance, and Beyond.Front Endocrinol (Lausanne). 2019 Jul 23;10:490. doi: 10.3389/fendo.2019.00490. eCollection 2019. Front Endocrinol (Lausanne). 2019. PMID: 31428045 Free PMC article. Review.
-
S1P-S1PR1 Signaling: the "Sphinx" in Osteoimmunology.Front Immunol. 2019 Jun 25;10:1409. doi: 10.3389/fimmu.2019.01409. eCollection 2019. Front Immunol. 2019. PMID: 31293578 Free PMC article. Review.
-
NF-κB signaling participates in both RANKL- and IL-4-induced macrophage fusion: receptor cross-talk leads to alterations in NF-κB pathways.J Immunol. 2011 Aug 15;187(4):1797-806. doi: 10.4049/jimmunol.1002628. Epub 2011 Jul 6. J Immunol. 2011. PMID: 21734075 Free PMC article.
-
Osteoimmunology: The Regulatory Roles of T Lymphocytes in Osteoporosis.Front Endocrinol (Lausanne). 2020 Aug 11;11:465. doi: 10.3389/fendo.2020.00465. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 32849268 Free PMC article. Review.
-
Transplanted interleukin-4--secreting mesenchymal stromal cells show extended survival and increased bone mineral density in the murine femur.Cytotherapy. 2018 Aug;20(8):1028-1036. doi: 10.1016/j.jcyt.2018.06.009. Epub 2018 Aug 2. Cytotherapy. 2018. PMID: 30077567 Free PMC article.
References
-
- Teitelbaum SL, Abu-Amer Y, Ross FP. Molecular mechanisms of bone resorption. J Cell Biochem. 1995;59:1–10. - PubMed
-
- Teitelbaum SL, Tondravi MM, Ross FP. Osteoclasts, macrophages, and the molecular mechanisms of bone resorption. J Leukoc Biol. 1997;61:381–388. - PubMed
-
- Takahashi N, et al. Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology. 1988;122:1373–1382. - PubMed
-
- Udagawa N, et al. The bone marrow-derived stromal cell lines MC3T3-G2/PA6 and ST2 support osteoclast-like cell differentiation in cocultures with mouse spleen cells. Endocrinology. 1989;125:1805–1813. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
