

Identification of TFII-I as the endoplasmic reticulum stress response element binding factor ERSF: its autoregulation by stress and interaction with ATF6
- PMID: 11287625
- PMCID: PMC86961
- DOI: 10.1128/MCB.21.9.3220-3233.2001
Identification of TFII-I as the endoplasmic reticulum stress response element binding factor ERSF: its autoregulation by stress and interaction with ATF6
Abstract
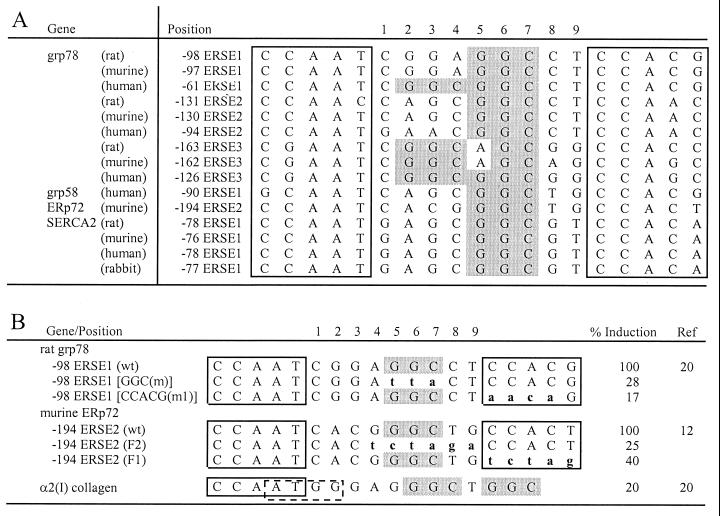
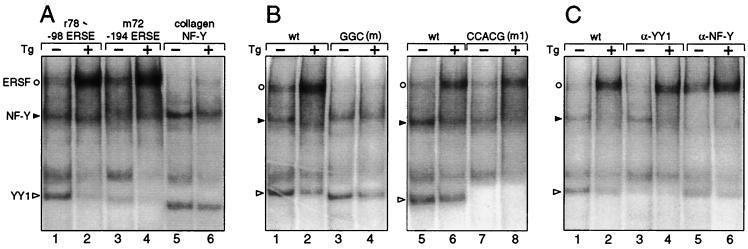
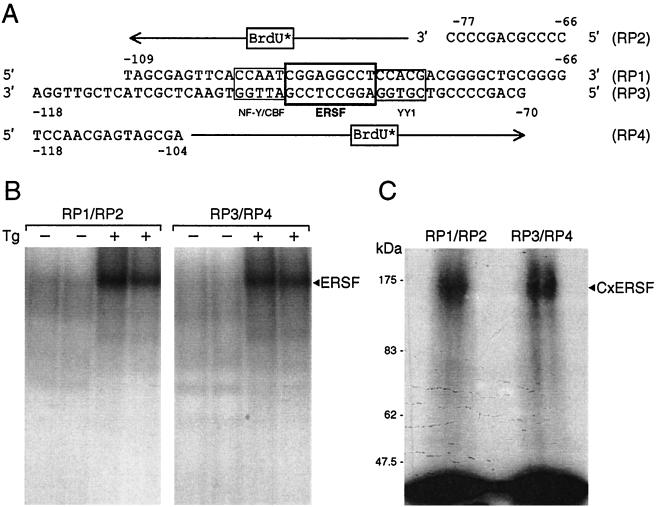
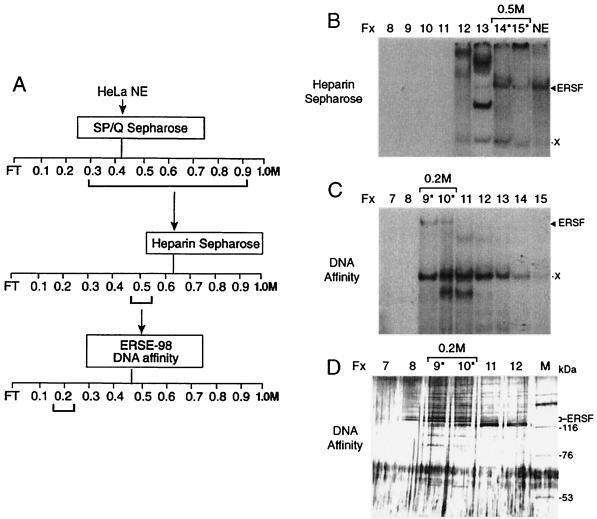
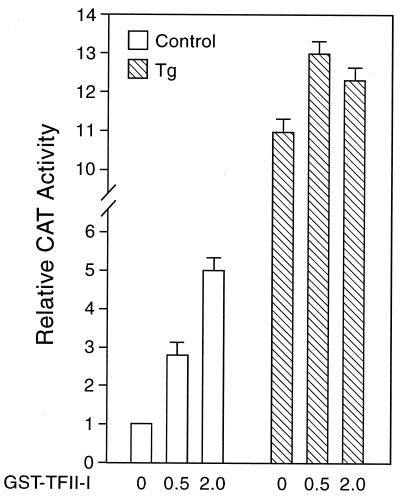
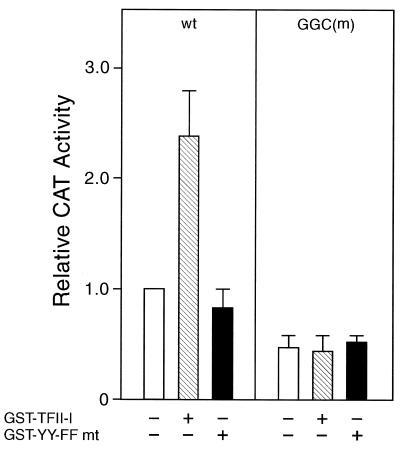
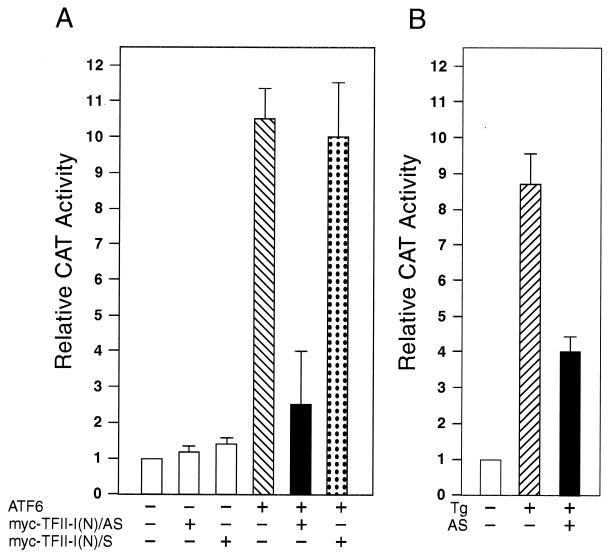
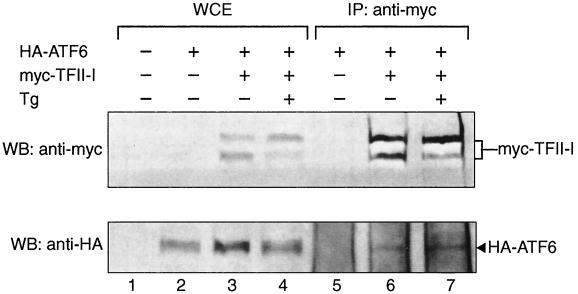
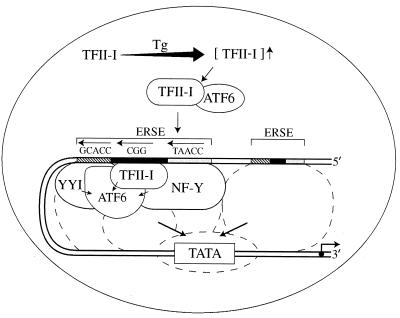
When mammalian cells are subjected to stress targeted to the endoplasmic reticulum (ER), such as depletion of the ER Ca(2+) store, the transcription of a family of glucose-regulated protein (GRP) genes encoding ER chaperones is induced. The GRP promoters contain multiple copies of the ER stress response element (ERSE), consisting of a unique tripartite structure, CCAAT(N(9))CCACG. Within a subset of mammalian ERSEs, N(9) represents a GC-rich sequence of 9 bp that is conserved across species. A novel complex (termed ERSF) exhibits enhanced binding to the ERSE of the grp78 and ERp72 promoters using HeLa nuclear extracts prepared from ER-stressed cells. Optimal binding of ERSF to ERSE and maximal ERSE-mediated stress inducibility require the conserved GGC motif within the 9-bp region. Through chromatographic purification and subsequent microsequencing, we have identified ERSF as TFII-I. Whereas TFII-I remains predominantly nuclear in both nontreated NIH 3T3 cells and cells treated with thapsigargin (Tg), a potent inducer of the GRP stress response through depletion of the ER Ca(2+) store, the level of TFII-I transcript was elevated in Tg-stressed cells, correlating with an increase in TFII-I protein level in the nuclei of Tg-stressed cells. Purified recombinant TFII-I isoforms bind directly to the ERSEs of grp78 and ERp72 promoters. The stimulation of ERSE-mediated transcription by TFII-I requires the consensus tyrosine phosphorylation site of TFII-I and the GGC sequence motif of the ERSE. We further discovered that TFII-I is an interactive protein partner of ATF6 and that optimal stimulation of ERSE by ATF6 requires TFII-I.
Figures

Similar articles
-
The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex.Nucleic Acids Res. 1999 Mar 15;27(6):1437-43. doi: 10.1093/nar/27.6.1437. Nucleic Acids Res. 1999. PMID: 10037803 Free PMC article.
-
ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1.Mol Cell Biol. 2000 Jul;20(14):5096-106. doi: 10.1128/MCB.20.14.5096-5106.2000. Mol Cell Biol. 2000. PMID: 10866666 Free PMC article.
-
Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response.Mol Cell Biol. 2001 Feb;21(4):1239-48. doi: 10.1128/MCB.21.4.1239-1248.2001. Mol Cell Biol. 2001. PMID: 11158310 Free PMC article.
-
The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications.Crit Rev Eukaryot Gene Expr. 1994;4(1):1-18. doi: 10.1615/critreveukargeneexpr.v4.i1.10. Crit Rev Eukaryot Gene Expr. 1994. PMID: 7987045 Review.
-
BiP binding keeps ATF6 at bay.Dev Cell. 2002 Jul;3(1):1-2. doi: 10.1016/s1534-5807(02)00210-1. Dev Cell. 2002. PMID: 12110159 Review.
Cited by
-
Transcriptional activation of endoplasmic reticulum chaperone GRP78 by HCMV IE1-72 protein.Cell Res. 2011 Apr;21(4):642-53. doi: 10.1038/cr.2011.10. Epub 2011 Jan 11. Cell Res. 2011. PMID: 21221131 Free PMC article.
-
Loss of cell surface TFII-I promotes apoptosis in prostate cancer cells stimulated with activated α₂ -macroglobulin.J Cell Biochem. 2011 Jun;112(6):1685-95. doi: 10.1002/jcb.23083. J Cell Biochem. 2011. PMID: 21503958 Free PMC article.
-
Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein.Proc Natl Acad Sci U S A. 2011 May 10;108(19):7832-7. doi: 10.1073/pnas.1101379108. Epub 2011 Apr 26. Proc Natl Acad Sci U S A. 2011. PMID: 21521793 Free PMC article.
-
Binding of Gtf2i-β/δ transcription factors to the ARMS2 gene leads to increased circulating HTRA1 in AMD patients and in vitro.J Biol Chem. 2021 Jan-Jun;296:100456. doi: 10.1016/j.jbc.2021.100456. Epub 2021 Feb 24. J Biol Chem. 2021. PMID: 33636181 Free PMC article.
-
Dermatan Sulfate Is a Potential Regulator of IgH via Interactions With Pre-BCR, GTF2I, and BiP ER Complex in Pre-B Lymphoblasts.Front Immunol. 2021 May 25;12:680212. doi: 10.3389/fimmu.2021.680212. eCollection 2021. Front Immunol. 2021. PMID: 34113352 Free PMC article.
References
-
- Caspersen C, Pedersen P S, Treiman M. The sarco/endoplasmic reticulum calcium-ATPase 2b is an endoplasmic reticulum stress-inducible protein. J Biol Chem. 2000;275:22363–22372. - PubMed
-
- Cheriyath V, Roy A L. Alternatively spliced isoforms of TFII-I. Complex formation, nuclear translocation, and differential gene regulation. J Biol Chem. 2000;275:26300–26308. - PubMed
-
- Foti D M, Welihinda A, Kaufman R J, Lee A S. Conservation and divergence of the yeast and mammalian unfolded protein response. Activation of specific mammalian endoplasmic reticulum stress element of the grp78/BiP promoter by yeast Hac1. J Biol Chem. 1999;274:30402–30409. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous