

Insulin inhibits transcription of IRS-2 gene in rat liver through an insulin response element (IRE) that resembles IREs of other insulin-repressed genes
- PMID: 11259670
- PMCID: PMC31125
- DOI: 10.1073/pnas.071054598
Insulin inhibits transcription of IRS-2 gene in rat liver through an insulin response element (IRE) that resembles IREs of other insulin-repressed genes
Abstract
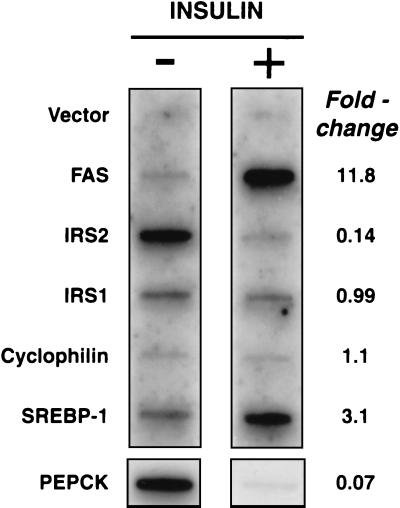
Recent data indicate that sustained elevations in plasma insulin suppress the mRNA for IRS-2, a component of the insulin signaling pathway in liver, and that this deficiency contributes to hepatic insulin resistance and inappropriate gluconeogenesis. Here, we use nuclear run-on assays to show that insulin inhibits transcription of the IRS-2 gene in the livers of intact rats. Insulin also inhibited transcription of a reporter gene driven by the human IRS-2 promoter that was transfected into freshly isolated rat hepatocytes. The human promoter contains a heptanucleotide sequence, TGTTTTG, that is identical to the insulin response element (IRE) identified previously in the promoters of insulin-repressed genes. Single base pair substitutions in this IRE decreased transcription of the IRS-2-driven reporter in the absence of insulin and abolished insulin-mediated repression. We conclude that insulin represses transcription of the IRS-2 gene by blocking the action of a positive factor that binds to the IRE. Sustained repression of IRS-2, as occurs in chronic hyperinsulinemia, contributes to hepatic insulin resistance and accelerates the development of the diabetic state.
Figures




Similar articles
-
SREBPs suppress IRS-2-mediated insulin signalling in the liver.Nat Cell Biol. 2004 Apr;6(4):351-7. doi: 10.1038/ncb1111. Epub 2004 Mar 14. Nat Cell Biol. 2004. PMID: 15048126
-
Characterization and regulation of the mouse insulin receptor substrate gene promoter.Mol Endocrinol. 1995 Oct;9(10):1367-79. doi: 10.1210/mend.9.10.8544845. Mol Endocrinol. 1995. PMID: 8544845
-
Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice.Mol Cell. 2000 Jul;6(1):77-86. Mol Cell. 2000. PMID: 10949029
-
FoxO feedback control of basal IRS-2 expression in pancreatic β-cells is distinct from that in hepatocytes.Diabetes. 2011 Nov;60(11):2883-91. doi: 10.2337/db11-0340. Epub 2011 Sep 20. Diabetes. 2011. PMID: 21933986 Free PMC article.
-
[Characterization of the IRS-1 (insulin receptor substrate-1) gene and its promoter].Nihon Rinsho. 1994 Oct;52(10):2659-64. Nihon Rinsho. 1994. PMID: 7983795 Review. Japanese.
Cited by
-
Gymnema Sylvestre Supplementation Restores Normoglycemia, Corrects Dyslipidemia, and Transcriptionally Modulates Pancreatic and Hepatic Gene Expression in Alloxan-Induced Hyperglycemic Rats.Metabolites. 2023 Apr 4;13(4):516. doi: 10.3390/metabo13040516. Metabolites. 2023. PMID: 37110174 Free PMC article.
-
The double-stranded RNA-dependent protein kinase differentially regulates insulin receptor substrates 1 and 2 in HepG2 cells.Mol Biol Cell. 2010 Oct 1;21(19):3449-58. doi: 10.1091/mbc.E10-06-0481. Epub 2010 Aug 4. Mol Biol Cell. 2010. PMID: 20685959 Free PMC article.
-
Expression and function of the insulin receptor substrate proteins in cancer.Cell Commun Signal. 2009 Jun 17;7:14. doi: 10.1186/1478-811X-7-14. Cell Commun Signal. 2009. PMID: 19534786 Free PMC article.
-
Insulin-receptor substrate-2 (irs-2) is required for maintaining glucokinase and glucokinase regulatory protein expression in mouse liver.PLoS One. 2013;8(4):e58797. doi: 10.1371/journal.pone.0058797. Epub 2013 Apr 1. PLoS One. 2013. PMID: 23560040 Free PMC article.
-
Insulin Controls Clock Gene Expression in the Liver of Goldfish Probably via Pi3k/Akt Pathway.Int J Mol Sci. 2023 Jul 25;24(15):11897. doi: 10.3390/ijms241511897. Int J Mol Sci. 2023. PMID: 37569272 Free PMC article.
References
-
- Kahn C R. Diabetes. 1994;43:1066–1084. - PubMed
-
- Shimomura I, Matsuda M, Hammer R E, Bashmakov Y, Brown M S, Goldstein J L. Mol Cell. 2000;6:77–86. - PubMed
-
- White M F. Mol Cell Biochem. 1998;182:3–11. - PubMed
-
- Withers D J, Gutierrez J S, Towery H, Burks D J, Ren J-M, Previs S, Zhang Y, Bernal D, Pons S, Shulman G I, et al. Nature (London) 1998;391:900–904. - PubMed
-
- Previs S F, Withers D J, Ren J-M, White M F, Shulman G I. J Biol Chem. 2000;275:38990–38994. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
