

Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication
- PMID: 11222695
- PMCID: PMC115896
- DOI: 10.1128/JVI.75.6.2710-2728.2001
Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication
Retraction in
-
Retraction for Zachos et al., "Herpes Simplex Virus Type 1 Blocks the Apoptotic Host Cell Defense Mechanisms That Target Bcl-2 and Manipulates Activation of p38 Mitogen-Activated Protein Kinase to Improve Viral Replication".J Virol. 2022 Sep 28;96(18):e0104422. doi: 10.1128/jvi.01044-22. Epub 2022 Aug 30. J Virol. 2022. PMID: 36040177 Free PMC article. No abstract available.
Abstract
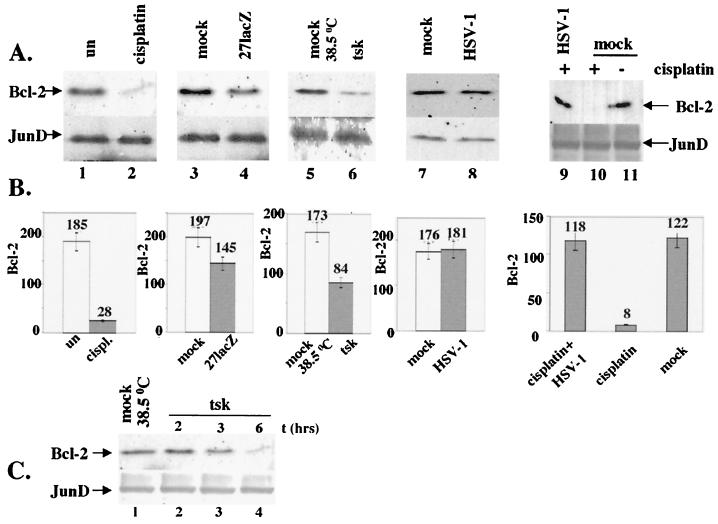
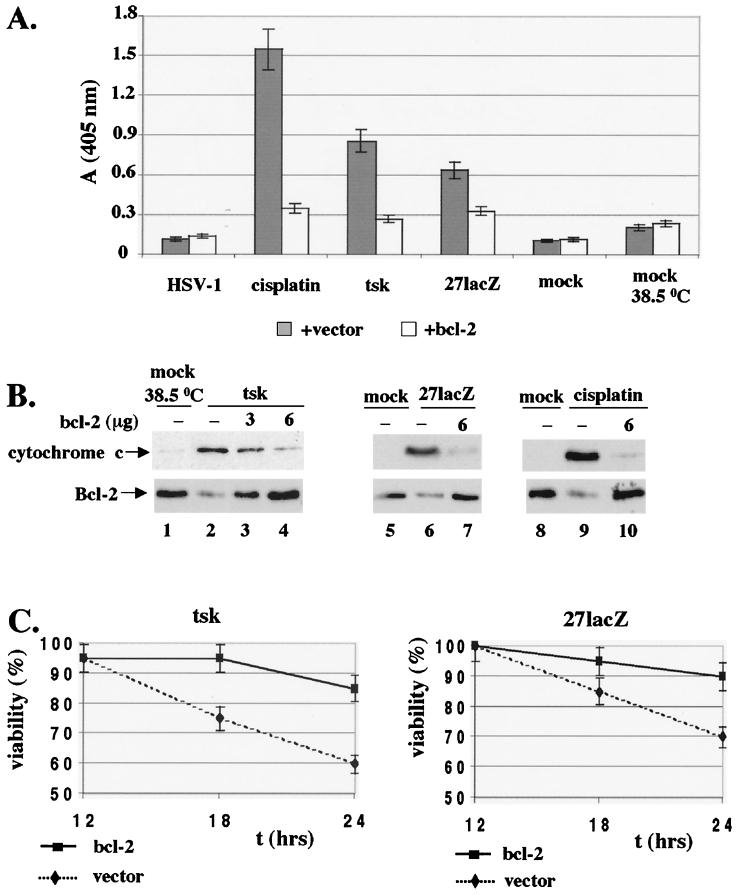
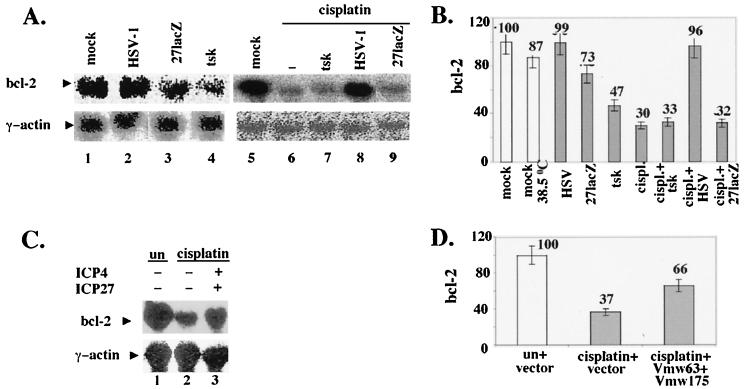
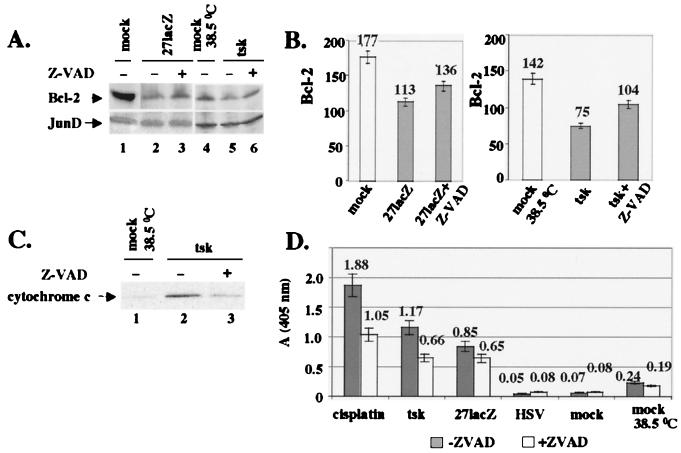
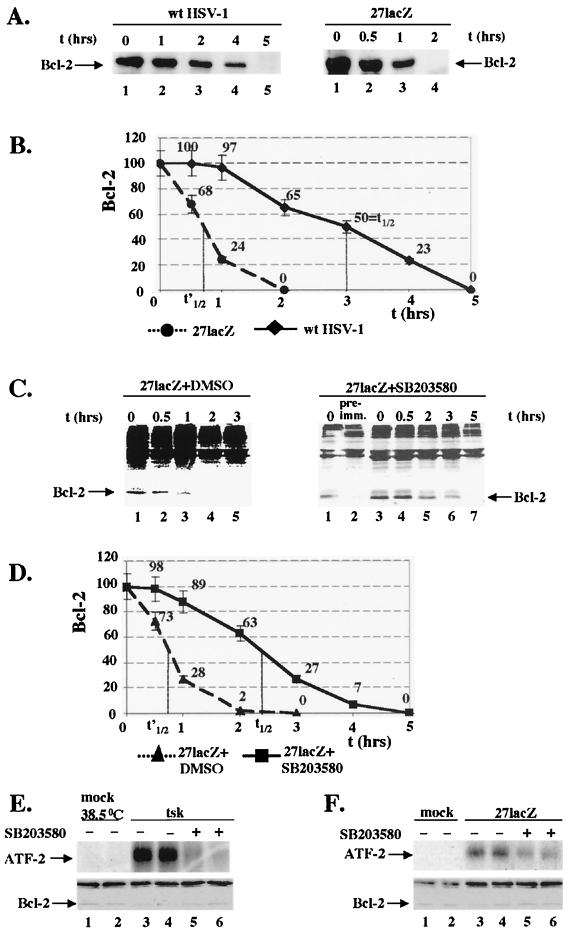
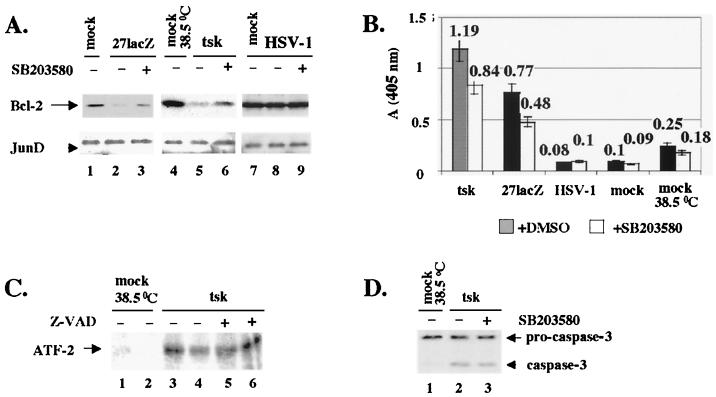
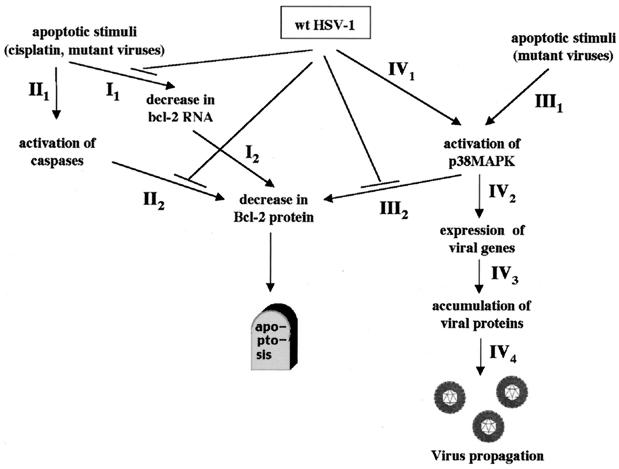
Wild-type (wt) herpes simplex virus type 1 (HSV-1) suppresses cell death. We investigated the apoptotic pathways triggered during infection with mutant viruses tsk and 27lacZ (which lack functional ICP4 and ICP27 viral proteins, respectively) and examined the mechanisms used by wt HSV-1 to protect against programmed cell death induced by the DNA-damaging compound cisplatin. In our studies, we used BHK and HeLa cells, with similar results. We suggest that a decrease in the levels of Bcl-2 protein is a key event during apoptosis induced by the mutant viruses and that Bcl-2 levels are targeted by (i) a decrease of bcl-2 RNA, (ii) caspase-related proteolysis, and (iii) p38 mitogen-activated protein kinase (p38MAPK)-dependent destabilization of Bcl-2 protein. We show that wt HSV-1, but not the mutant viruses, maintains bcl-2 RNA and protein levels during infection and protects from the cisplatin-induced decrease in bcl-2 RNA; our data suggest that both ICP27 and ICP4 are required for this function. Additionally, wt HSV-1 evades but does not actively block activation of caspases. Although wt HSV-1 induces p38MAPK activation during infection, it prevents p38MAPK-dependent destabilization of Bcl-2 and exploits p38MAPK stimulation to enhance transcription of specific viral gene promoters to increase viral yields.
Figures


Similar articles
-
Bcl-2 blocks a caspase-dependent pathway of apoptosis activated by herpes simplex virus 1 infection in HEp-2 cells.J Virol. 2000 Feb;74(4):1931-8. doi: 10.1128/jvi.74.4.1931-1938.2000. J Virol. 2000. PMID: 10644366 Free PMC article.
-
Herpes simplex virus type 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells.J Virol. 2009 Feb;83(4):1767-77. doi: 10.1128/JVI.01944-08. Epub 2008 Dec 10. J Virol. 2009. PMID: 19073744 Free PMC article.
-
c-Myc potentiates the mitochondrial pathway of apoptosis by acting upstream of apoptosis signal-regulating kinase 1 (Ask1) in the p38 signalling cascade.Biochem J. 2003 Jun 1;372(Pt 2):631-41. doi: 10.1042/BJ20021565. Biochem J. 2003. PMID: 12646044 Free PMC article.
-
The ATM and Rad3-Related (ATR) Protein Kinase Pathway Is Activated by Herpes Simplex Virus 1 and Required for Efficient Viral Replication.J Virol. 2018 Feb 26;92(6):e01884-17. doi: 10.1128/JVI.01884-17. Print 2018 Mar 15. J Virol. 2018. PMID: 29263259 Free PMC article.
-
Poly(A)-binding protein 1 partially relocalizes to the nucleus during herpes simplex virus type 1 infection in an ICP27-independent manner and does not inhibit virus replication.J Virol. 2010 Sep;84(17):8539-48. doi: 10.1128/JVI.00668-10. Epub 2010 Jun 23. J Virol. 2010. PMID: 20573819 Free PMC article.
Cited by
-
CK2 protein kinase is stimulated and redistributed by functional herpes simplex virus ICP27 protein.J Virol. 2003 Apr;77(7):4315-25. doi: 10.1128/jvi.77.7.4315-4325.2003. J Virol. 2003. PMID: 12634389 Free PMC article.
-
Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons.J Virol. 2006 Jan;80(2):1025-31. doi: 10.1128/JVI.80.2.1025-1031.2006. J Virol. 2006. PMID: 16379003 Free PMC article.
-
The suppression of apoptosis by α-herpesvirus.Cell Death Dis. 2017 Apr 13;8(4):e2749. doi: 10.1038/cddis.2017.139. Cell Death Dis. 2017. PMID: 28406478 Free PMC article. Review.
-
Identification of differentially activated cell-signaling networks associated with pichinde virus pathogenesis by using systems kinomics.J Virol. 2007 Feb;81(4):1923-33. doi: 10.1128/JVI.02199-06. Epub 2006 Dec 6. J Virol. 2007. PMID: 17151108 Free PMC article.
-
Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response.J Virol. 2002 May;76(9):4162-71. doi: 10.1128/jvi.76.9.4162-4171.2002. J Virol. 2002. PMID: 11932381 Free PMC article.
References
-
- Adams J M, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. - PubMed
-
- Allen K E, Everett R D. Mutations which alter the DNA binding properties of the herpes simplex virus type 1 transactivating protein Vmw175 also affect its ability to support virus replication. J Gen Virol. 1997;78:2913–2922. - PubMed
-
- Conner J, Murray J, Cross A, Clements J B, Marsden H S. Intracellular localisation of herpes simplex virus type 1 ribonucleotide reductase subunits during infection of cultured cells. Virology. 1995;213:615–623. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
