

A reversible component of mitochondrial respiratory dysfunction in apoptosis can be rescued by exogenous cytochrome c
- PMID: 11179211
- PMCID: PMC145422
- DOI: 10.1093/emboj/20.4.661
A reversible component of mitochondrial respiratory dysfunction in apoptosis can be rescued by exogenous cytochrome c
Abstract
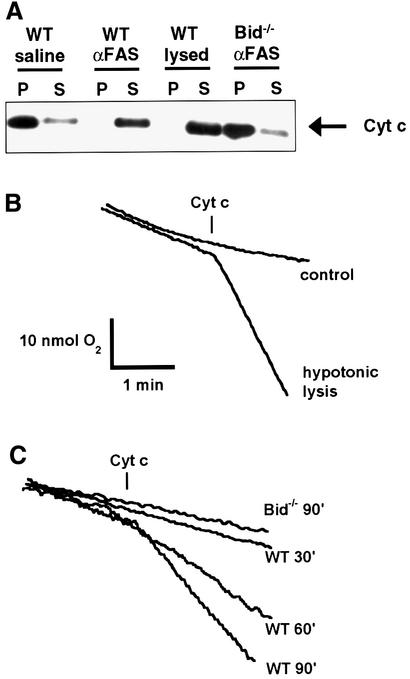
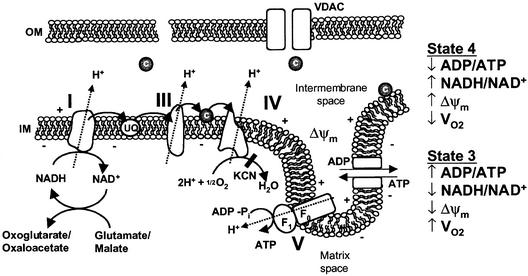
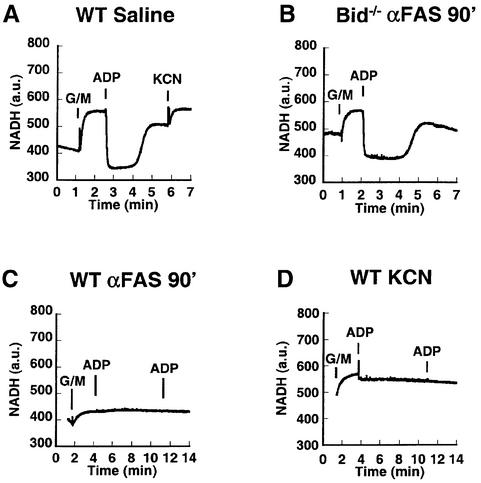
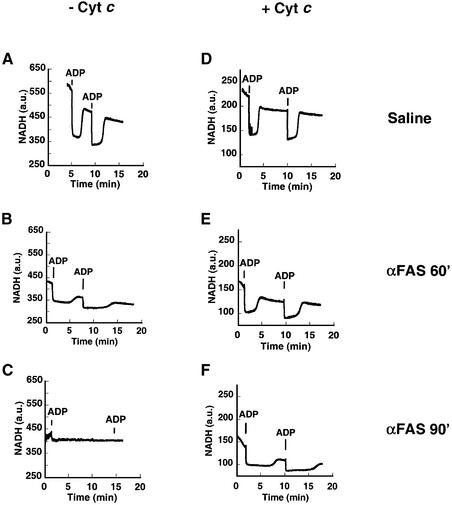
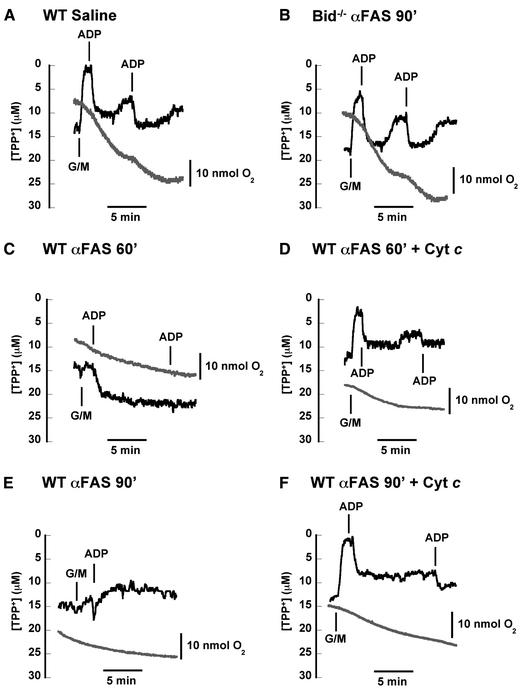
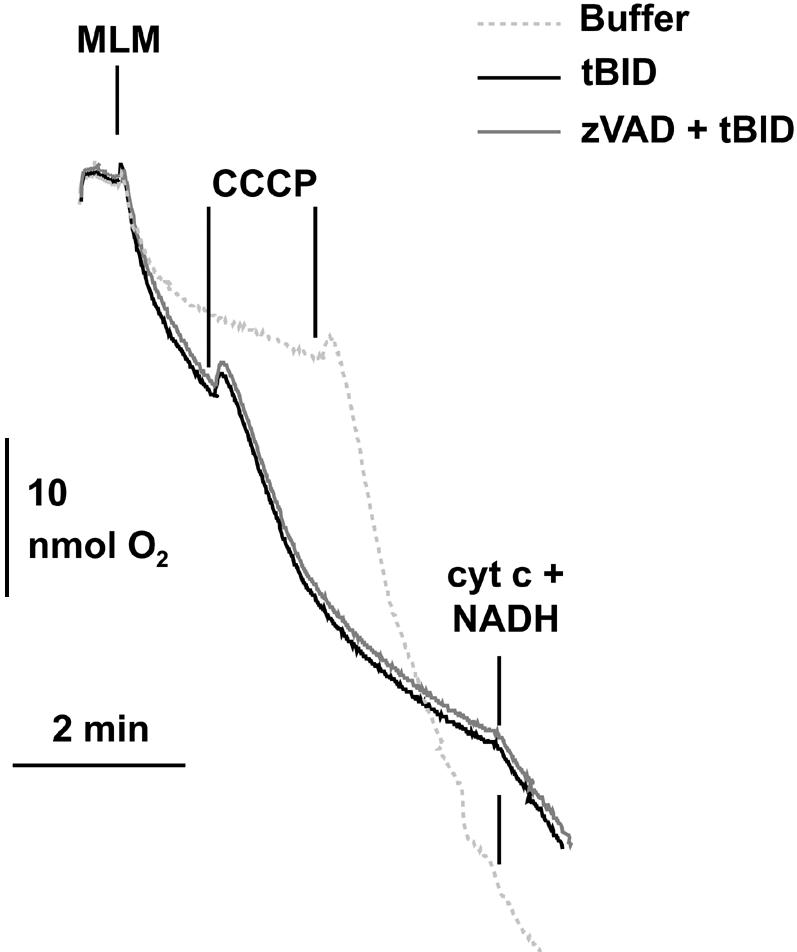
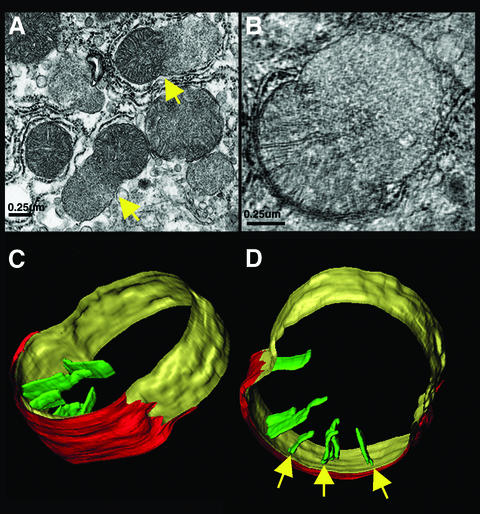
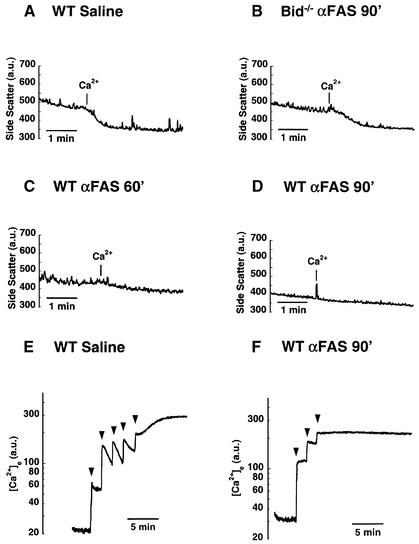
Multiple apoptotic pathways release cytochrome c from the mitochondrial intermembrane space, resulting in the activation of downstream caspases. In vivo activation of Fas (CD95) resulted in increased permeability of the mitochondrial outer membrane and depletion of cytochrome c stores. Serial measurements of oxygen consumption, NADH redox state and membrane potential revealed a loss of respiratory state transitions. This tBID-induced respiratory failure did not require any caspase activity. At early time points, re-addition of exogenous cytochrome c markedly restored respiratory functions. Over time, however, mitochondria showed increasing irreversible respiratory dysfunction as well as diminished calcium buffering. Electron microscopy and tomographic reconstruction revealed asymmetric mitochondria with blebs of herniated matrix, distended inner membrane and partial loss of cristae structure. Thus, apoptogenic redistribution of cytochrome c is responsible for a distinct program of mitochondrial respiratory dysfunction, in addition to the activation of downstream caspases.
Figures
Similar articles
-
Bid-induced cytochrome c release is mediated by a pathway independent of mitochondrial permeability transition pore and Bax.J Biol Chem. 2000 Dec 15;275(50):39474-81. doi: 10.1074/jbc.M003370200. J Biol Chem. 2000. PMID: 10982793
-
Rate-limiting step preceding cytochrome c release in cells primed for Fas-mediated apoptosis revealed by analysis of cellular mosaicism of respiratory changes.J Biol Chem. 2001 Jan 5;276(1):606-15. doi: 10.1074/jbc.M007871200. J Biol Chem. 2001. PMID: 11027691
-
Uncouplers of oxidative phosphorylation can enhance a Fas death signal.Mol Cell Biol. 1999 May;19(5):3299-311. doi: 10.1128/MCB.19.5.3299. Mol Cell Biol. 1999. PMID: 10207055 Free PMC article.
-
Fas death receptor signalling: roles of Bid and XIAP.Cell Death Differ. 2012 Jan;19(1):42-50. doi: 10.1038/cdd.2011.121. Epub 2011 Sep 30. Cell Death Differ. 2012. PMID: 21959933 Free PMC article. Review.
-
Mitochondrial control of apoptosis: the role of cytochrome c.Biochim Biophys Acta. 1998 Aug 10;1366(1-2):139-49. doi: 10.1016/s0005-2728(98)00109-1. Biochim Biophys Acta. 1998. PMID: 9714780 Review.
Cited by
-
Exposure of the inner mitochondrial membrane triggers apoptotic mitophagy.Cell Death Differ. 2024 Mar;31(3):335-347. doi: 10.1038/s41418-024-01260-2. Epub 2024 Feb 23. Cell Death Differ. 2024. PMID: 38396150 Free PMC article.
-
Mitochondrial function in apoptotic neuronal cell death.Neurochem Res. 2004 Mar;29(3):521-30. doi: 10.1023/b:nere.0000014823.74782.b7. Neurochem Res. 2004. PMID: 15038600 Review.
-
Mitochondrial longevity pathways.Biochim Biophys Acta. 2011 Apr;1813(4):634-44. doi: 10.1016/j.bbamcr.2011.01.029. Epub 2011 Feb 2. Biochim Biophys Acta. 2011. PMID: 21295080 Free PMC article. Review.
-
The caspase pathway of linoelaidic acid (9t, 12t-c18:2)-induced apoptosis in human umbilical vein endothelial cells.Lipids. 2013 Feb;48(2):115-26. doi: 10.1007/s11745-012-3728-4. Epub 2012 Oct 13. Lipids. 2013. PMID: 23065354
-
Regulation of cell death by transfer RNA.Antioxid Redox Signal. 2013 Aug 20;19(6):583-94. doi: 10.1089/ars.2012.5171. Epub 2013 Mar 28. Antioxid Redox Signal. 2013. PMID: 23350625 Free PMC article. Review.
References
-
- Amarante-Mendes G.P., Finucane,D.M., Martin,S.J., Cotter,T.G., Salvesen,G.S. and Green,D.R. (1998) Anti-apoptotic oncogenes prevent caspase-dependent and independent commitment for cell death. Cell Death Differ., 5, 298–306. - PubMed
-
- Baffy G., Miyashita,T., Williamson,J.R. and Reed,J.C. (1993) Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J. Biol. Chem., 268, 6511–6519. - PubMed
-
- Basanez M.G., Nechushtan,A., Drozhinin,O., Chanturiya,A., Choe,E., Tutt,S., Wood,K.A., Hsu,Y., Zimmerberg,J. and Youle,R.J. (1999) Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc. Natl Acad. Sci. USA, 96, 5492–5497. - PMC - PubMed
-
- Bernardi P. (1999) Mitochondrial transport of cations: channels, exchangers and permeability transition. Physiol. Rev., 79, 1127–1155. - PubMed
-
- Cai J. and Jones,D.P. (1998) Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem., 273, 11401–11404. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
