

Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1
- PMID: 11027346
- PMCID: PMC17223
- DOI: 10.1073/pnas.97.21.11466
Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1
Abstract
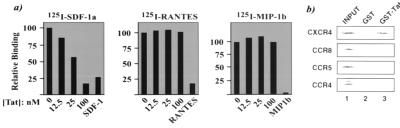
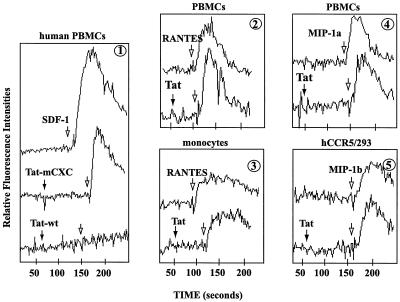
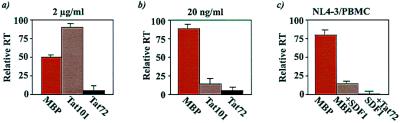
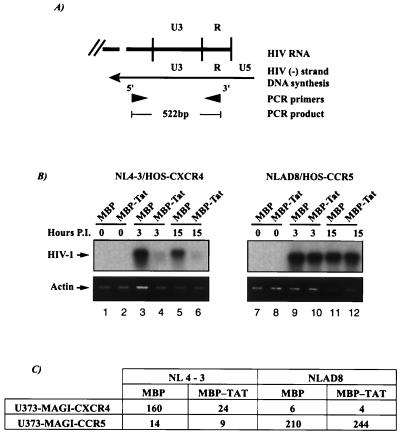
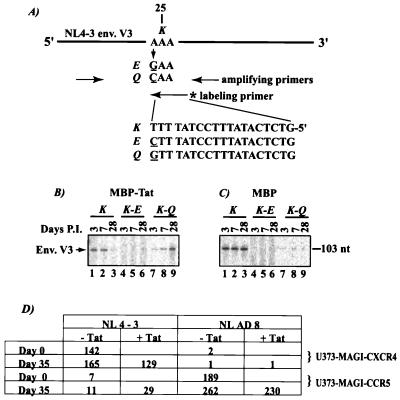
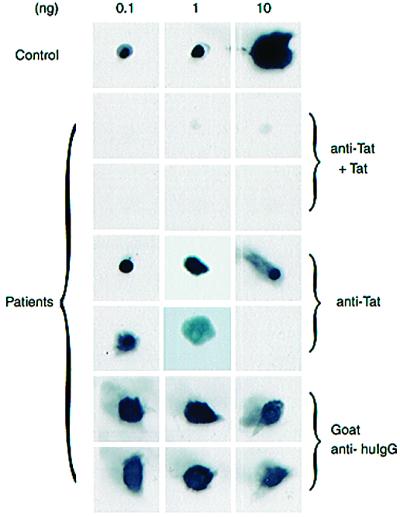
Chemokines and chemokine receptors play important roles in HIV-1 infection and tropism. CCR5 is the major macrophage-tropic coreceptor for HIV-1 whereas CXC chemokine receptor 4 (CXCR4) serves the counterpart function for T cell-tropic viruses. An outstanding biological mystery is why only R5-HIV-1 is initially detected in new seroconvertors who are exposed to R5 and X4 viruses. Indeed, X4 virus emerges in a minority of patients and only in the late stage of disease, suggesting that early negative selection against HIV-1-CXCR4 interaction may exist. Here, we report that the HIV-1 Tat protein, which is secreted from virus-infected cells, is a CXCR4-specific antagonist. Soluble Tat selectively inhibited the entry and replication of X4, but not R5, virus in peripheral blood mononuclear cells (PBMCs). We propose that one functional consequence of secreted Tat is to select against X4 viruses, thereby influencing the early in vivo course of HIV-1 disease.
Figures
Similar articles
-
Tat protein induces human immunodeficiency virus type 1 (HIV-1) coreceptors and promotes infection with both macrophage-tropic and T-lymphotropic HIV-1 strains.J Virol. 1998 Nov;72(11):8952-60. doi: 10.1128/JVI.72.11.8952-8960.1998. J Virol. 1998. PMID: 9765440 Free PMC article.
-
Dual-tropic HIV type 1 isolates vary dramatically in their utilization of CCR5 and CXCR4 coreceptors.AIDS. 2010 Sep 10;24(14):2181-6. doi: 10.1097/QAD.0b013e32833c543f. AIDS. 2010. PMID: 20639728
-
Differences in molecular evolution between switch (R5 to R5X4/X4-tropic) and non-switch (R5-tropic only) HIV-1 populations during infection.Infect Genet Evol. 2010 Apr;10(3):356-64. doi: 10.1016/j.meegid.2009.05.003. Epub 2009 May 14. Infect Genet Evol. 2010. PMID: 19446658
-
HIV co-receptors as targets for antiviral therapy.Curr Top Med Chem. 2004;4(9):883-93. doi: 10.2174/1568026043388501. Curr Top Med Chem. 2004. PMID: 15134547 Review.
-
Effect of HIV-1 subtype and tropism on treatment with chemokine coreceptor entry inhibitors; overview of viral entry inhibition.Crit Rev Microbiol. 2015;41(4):473-87. doi: 10.3109/1040841X.2013.867829. Epub 2014 Mar 17. Crit Rev Microbiol. 2015. PMID: 24635642 Review.
Cited by
-
Anti-Tat immunity defines CD4+ T-cell dynamics in people living with HIV on long-term cART.EBioMedicine. 2021 Apr;66:103306. doi: 10.1016/j.ebiom.2021.103306. Epub 2021 Apr 7. EBioMedicine. 2021. PMID: 33839064 Free PMC article.
-
HIV-1 Tat Protein Activates both the MyD88 and TRIF Pathways To Induce Tumor Necrosis Factor Alpha and Interleukin-10 in Human Monocytes.J Virol. 2016 Jun 10;90(13):5886-5898. doi: 10.1128/JVI.00262-16. Print 2016 Jul 1. J Virol. 2016. PMID: 27053552 Free PMC article.
-
Role of HIV-1 Tat Protein Interactions with Host Receptors in HIV Infection and Pathogenesis.Int J Mol Sci. 2024 Jan 30;25(3):1704. doi: 10.3390/ijms25031704. Int J Mol Sci. 2024. PMID: 38338977 Free PMC article. Review.
-
Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline.Am J Pathol. 2003 May;162(5):1693-707. doi: 10.1016/S0002-9440(10)64304-0. Am J Pathol. 2003. PMID: 12707054 Free PMC article.
-
High-performance capillary electrophoresis for determining HIV-1 Tat protein in neurons.PLoS One. 2011 Jan 7;6(1):e16148. doi: 10.1371/journal.pone.0016148. PLoS One. 2011. PMID: 21249135 Free PMC article.
References
-
- Berger E A, Murphy P M, Farber J M. Annu Rev Immunol. 1999;17:657–700. - PubMed
-
- Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. - PubMed
-
- Deng H K, Unutmaz D, KewalRamani V N, Littman D R. Nature (London) 1996;381:661–666. - PubMed
-
- Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. Nature (London) 1996;381:667–673. - PubMed
-
- Berger E A, Doms R W, Fenyo E M, Korber B T, Littman D R, Moore J P, Sattentau Q J, Schuitemaker H, Sodroski J, Weiss R A. Nature (London) 1998;391:240. (lett.). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
