

The exceptionally large genome of Hendra virus: support for creation of a new genus within the family Paramyxoviridae
- PMID: 11024125
- PMCID: PMC102035
- DOI: 10.1128/jvi.74.21.9972-9979.2000
The exceptionally large genome of Hendra virus: support for creation of a new genus within the family Paramyxoviridae
Abstract
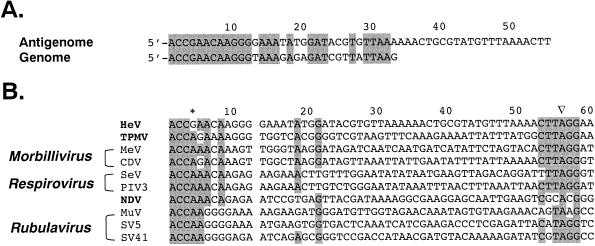
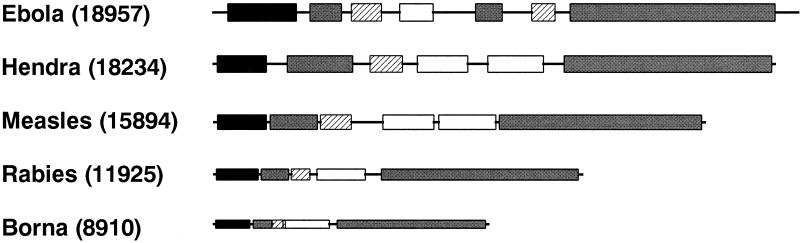
An outbreak of acute respiratory disease in Hendra, a suburb of Brisbane, Australia, in September 1994 resulted in the deaths of 14 racing horses and a horse trainer. The causative agent was a new member of the family Paramyxoviridae. The virus was originally called Equine morbillivirus but was renamed Hendra virus (HeV) when molecular characterization highlighted differences between it and members of the genus Morbillivirus. Less than 5 years later, the closely related Nipah virus (NiV) emerged in Malaysia, spread rapidly through the pig population, and caused the deaths of over 100 people. We report the characterization of the HeV L gene and protein, the genome termini, and gene boundary sequences, thus completing the HeV genome sequence. In the highly conserved region of the L protein, the HeV sequence GDNE differs from the GDNQ found in almost all other nonsegmented negative-strand (NNS) RNA viruses. HeV has an absolutely conserved intergenic trinucleotide sequence, 3'-GAA-5', and highly conserved transcription initiation and termination sequences similar to those of respiroviruses and morbilliviruses. The large genome size (18,234 nucleotides), the unique complementary genome terminal sequences of HeV, and the limited homology with other members of the Paramyxoviridae suggest that HeV, together with NiV, should be classified in a new genus in this family. The large genome of HeV also fills a gap in the spectrum of genome sizes observed with NNS RNA virus genomes. As such, it provides a further piece in the puzzle of NNS RNA virus evolution.
Figures

Similar articles
-
Molecular characterization of the polymerase gene and genomic termini of Nipah virus.Virology. 2001 Aug 15;287(1):192-201. doi: 10.1006/viro.2001.1026. Virology. 2001. PMID: 11504554
-
Sequence analysis of the Hendra virus nucleoprotein gene: comparison with other members of the subfamily Paramyxovirinae.J Gen Virol. 1998 Jul;79 ( Pt 7):1775-80. doi: 10.1099/0022-1317-79-7-1775. J Gen Virol. 1998. PMID: 9680142
-
Genome characterization of Salem virus reveals its evolutionary intermediate status in the subfamily Paramyxovirinae.Arch Virol. 2012 Oct;157(10):1989-93. doi: 10.1007/s00705-012-1388-6. Epub 2012 Jun 23. Arch Virol. 2012. PMID: 22729563
-
Molecular biology of Hendra and Nipah viruses.Microbes Infect. 2001 Apr;3(4):279-87. doi: 10.1016/s1286-4579(01)01381-8. Microbes Infect. 2001. PMID: 11334745 Review.
-
Hendra (equine morbillivirus).Vet J. 2000 Nov;160(3):169-76. doi: 10.1053/tvjl.2000.0508. Vet J. 2000. PMID: 11061954 Review.
Cited by
-
Emerging trends of Nipah virus: A review.Rev Med Virol. 2019 Jan;29(1):e2010. doi: 10.1002/rmv.2010. Epub 2018 Sep 24. Rev Med Virol. 2019. PMID: 30251294 Free PMC article. Review.
-
Assessment of the viral safety of antivenoms fractionated from equine plasma.Biologicals. 2004 Sep;32(3):115-28. doi: 10.1016/j.biologicals.2004.07.001. Biologicals. 2004. PMID: 15536042 Free PMC article. Review.
-
Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations.Arch Virol. 2010 Dec;155(12):2083-103. doi: 10.1007/s00705-010-0814-x. Epub 2010 Oct 30. Arch Virol. 2010. PMID: 21046175 Free PMC article.
-
Discovery of Potential Antiviral Compounds against Hendra Virus by Targeting Its Receptor-Binding Protein (G) Using Computational Approaches.Molecules. 2022 Jan 16;27(2):554. doi: 10.3390/molecules27020554. Molecules. 2022. PMID: 35056869 Free PMC article.
-
Role of endocytosis and cathepsin-mediated activation in Nipah virus entry.Virology. 2008 Jun 5;375(2):391-400. doi: 10.1016/j.virol.2008.02.019. Epub 2008 Mar 14. Virology. 2008. PMID: 18342904 Free PMC article.
References
-
- Allworth A, Murray K, Morgan J. A human case of encephalitis due to a lyssavirus recently identified in fruit bats. Commun Dis Intellig. 1996;20:504.
-
- Bellini W J, Rota P A, Anderson L J. Paramyxoviruses. In: Collier L, Balows A, Sussman M, editors. Microbiology and microbial infections. Vol. 1. London, England: Arnold; 1998. pp. 435–461.
-
- Chua K B, Bellini W J, Rota P A, Harcourt B H, Tamin A, Lam S K, Ksiazek T G, Rollin P E, Zaki S R, Shieh W J, Goldsmith C S, Roehrig J T, Eaton B, Gould A R, Olson J, Field H, Daniels P, Ling A E, Peters C J, Anderson L J, Mahy B W J. Nipah virus, a newly emergent deadly praramyxovirus. Science. 2000;288:1432–1435. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
