

Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1
- PMID: 10970861
- PMCID: PMC302047
- DOI: 10.1093/emboj/19.17.4688
Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1
Abstract
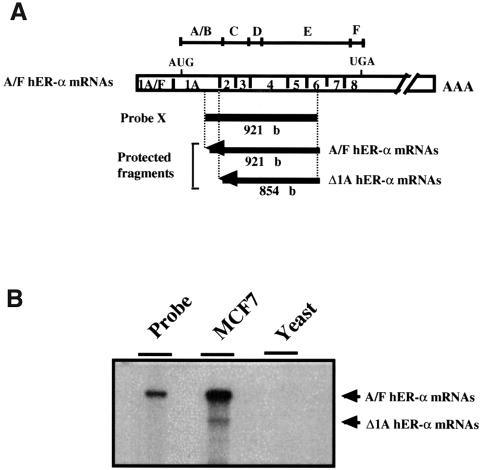
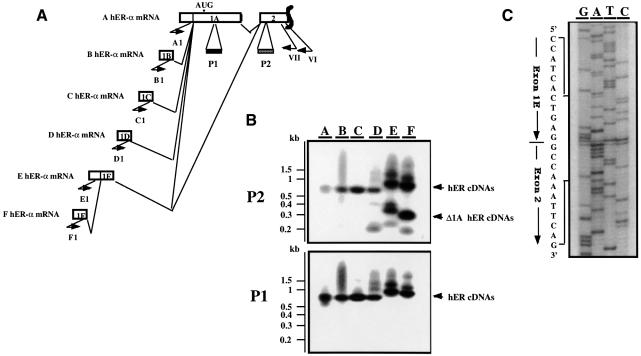
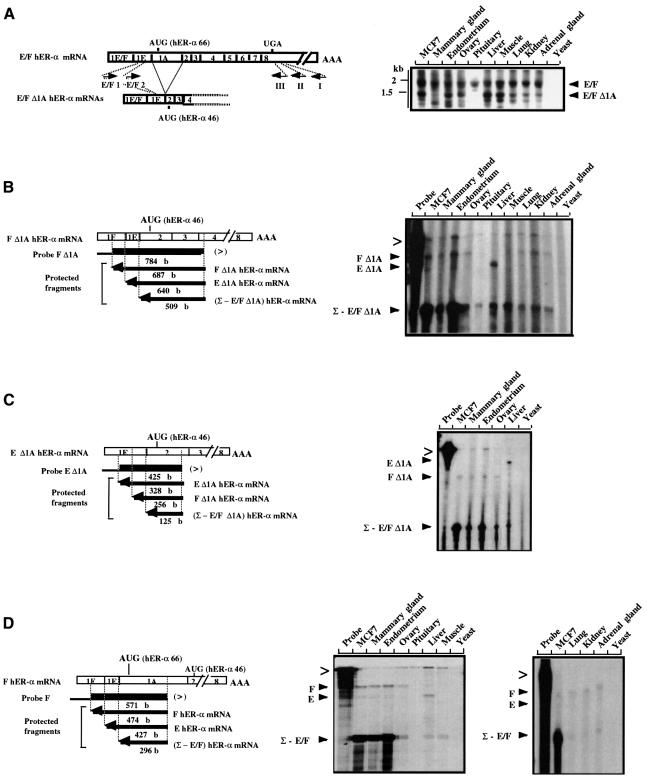
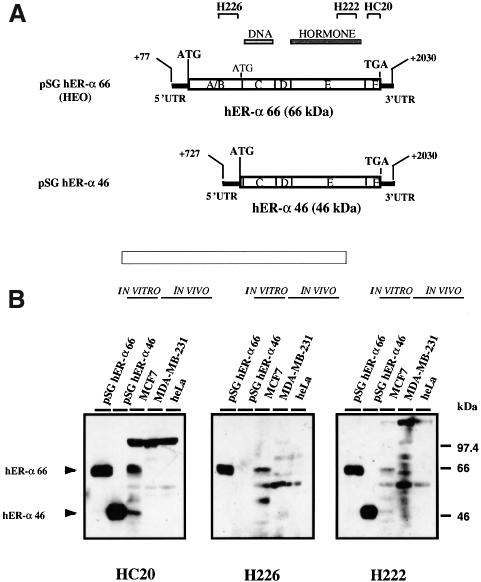
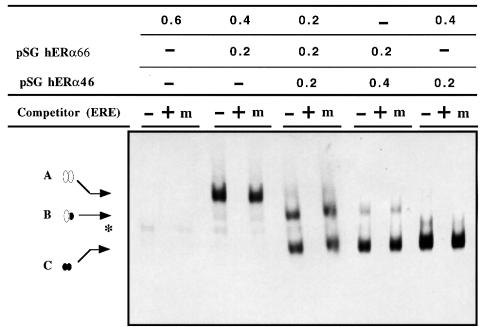
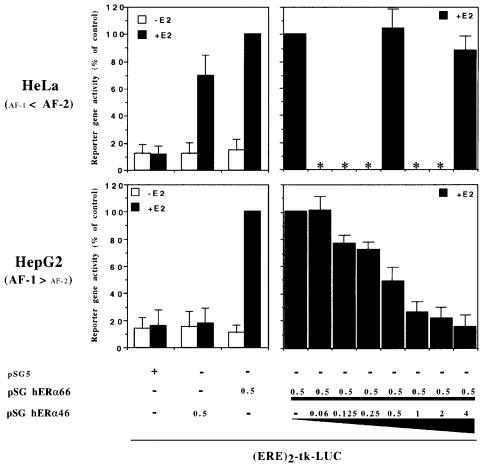
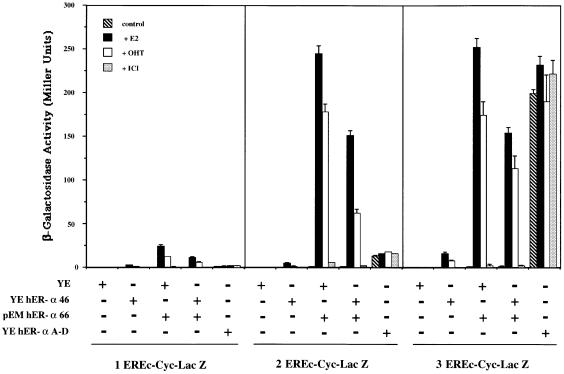
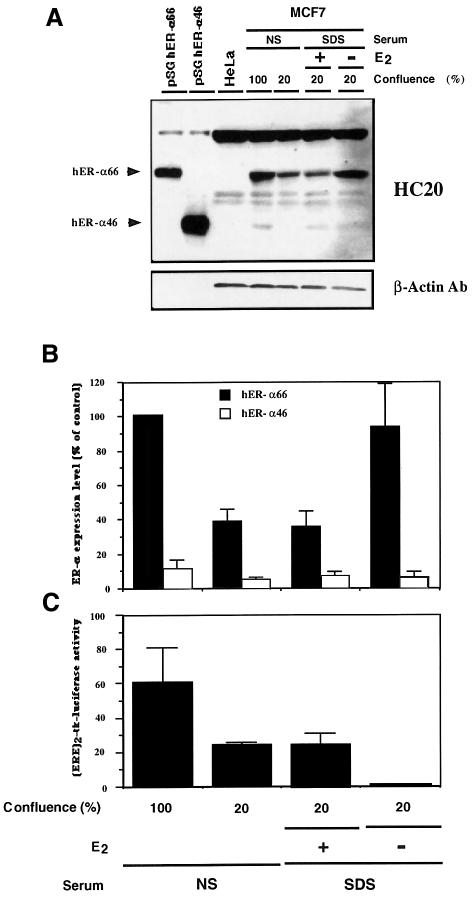
A new isoform of the human estrogen receptor-alpha (hER-alpha) has been identified and characterized. This 46 kDa isoform (hERalpha46) lacks the N-terminal 173 amino acids present in the previously characterized 66 kDa isoform (hERalpha66). hERalpha46 is encoded by a new class of hER-alpha transcript that lacks the first coding exon (exon 1A) of the ER-alpha gene. We demonstrated that these Delta1A hER-alpha transcripts originate from the E and F hER-alpha promoters and are produced by the splicing of exon 1E directly to exon 2. Functional analysis of hERalpha46 showed that, in a cell context sensitive to the transactivation function AF-2, this receptor is an effective ligand-inducible transcription factor. In contrast, hERalpha46 is a powerful inhibitor of hERalpha66 in a cell context where the transactivating function of AF-1 predominates over AF-2. The mechanisms by which the AF-1 dominant-negative action is exerted may involve heterodimeri zation of the two receptor isoforms and/or direct competition for the ER-alpha DNA-binding site. hERalpha66/hERalpha46 ratios change with the cell growth status of the breast carcinoma cell line MCF7, suggesting a role of hERalpha46 in cellular proliferation.
Figures
Similar articles
-
The human estrogen receptor-alpha isoform hERalpha46 antagonizes the proliferative influence of hERalpha66 in MCF7 breast cancer cells.Endocrinology. 2005 Dec;146(12):5474-84. doi: 10.1210/en.2005-0866. Epub 2005 Sep 8. Endocrinology. 2005. PMID: 16150902
-
ERalpha gene expression in human primary osteoblasts: evidence for the expression of two receptor proteins.Mol Endocrinol. 2001 Dec;15(12):2064-77. doi: 10.1210/mend.15.12.0741. Mol Endocrinol. 2001. PMID: 11731609
-
Two functionally different protein isoforms are produced from the chicken estrogen receptor-alpha gene.Mol Endocrinol. 1999 Sep;13(9):1571-87. doi: 10.1210/mend.13.9.0336. Mol Endocrinol. 1999. PMID: 10478847
-
Alternative splicing of the estrogen receptor primary transcript normally occurs in estrogen receptor positive tissues and cell lines.J Steroid Biochem Mol Biol. 1996 Jan;56(1-6 Spec No):99-105. doi: 10.1016/0960-0760(95)00227-8. J Steroid Biochem Mol Biol. 1996. PMID: 8603053 Review.
-
Steroid receptors and metastatic potential in endometrial cancers.J Steroid Biochem Mol Biol. 2000 Dec 31;75(4-5):209-12. doi: 10.1016/s0960-0760(00)00176-x. J Steroid Biochem Mol Biol. 2000. PMID: 11282273 Review.
Cited by
-
Divide and Conquer May Not Be the Optimal Approach to Retain the Desirable Estrogenic Attributes of the Cyclopia Nutraceutical Extract, SM6Met.PLoS One. 2015 Jul 24;10(7):e0132950. doi: 10.1371/journal.pone.0132950. eCollection 2015. PLoS One. 2015. PMID: 26208351 Free PMC article.
-
Aging and substitutive hormonal therapy influence in regional and subcellular distribution of ERα in female rat brain.Age (Dordr). 2013 Jun;35(3):821-37. doi: 10.1007/s11357-012-9415-9. Epub 2012 May 10. Age (Dordr). 2013. PMID: 22648398 Free PMC article.
-
Expression of the human oestrogen receptor-alpha gene is regulated by promoter F in MG-63 osteoblastic cells.Biochem J. 2003 Jun 15;372(Pt 3):831-9. doi: 10.1042/BJ20021633. Biochem J. 2003. PMID: 12659635 Free PMC article.
-
Alternative promoters and splicing create multiple functionally distinct isoforms of oestrogen receptor alpha in breast cancer and healthy tissues.Cancer Med. 2023 Sep;12(18):18931-18945. doi: 10.1002/cam4.6508. Epub 2023 Sep 7. Cancer Med. 2023. PMID: 37676103 Free PMC article.
-
Generation and characterization of a complete null estrogen receptor alpha mouse using Cre/LoxP technology.Mol Cell Biochem. 2009 Jan;321(1-2):145-53. doi: 10.1007/s11010-008-9928-9. Epub 2008 Oct 25. Mol Cell Biochem. 2009. PMID: 18953638 Free PMC article.
References
-
- Abbondanza C., De Falco,A., Nigro,V., Medici,N., Armetta,I., Molinari,A.M., Moncharmont,B. and Puca,G.A. (1993) Characterization and epitope mapping of a new panel of monoclonal antibodies to estradiol receptor. Steroids, 58, 4–12. - PubMed
-
- Auchus R.J. and Fuqua,S.A.W. (1994) The oestrogen receptor. In Bailliere’s Clinical Endocrinology and Metabolism: Hormones, Enzymes and Receptors, Vol. 8. Baillière Tindall, London, UK, pp. 433–449. - PubMed
-
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1989) Current Protocols in Molecular Biology. Wiley-Interscience, New York, NY.
-
- Banroques J., Delahodde,A. and Jacq,C. (1986) A mitochondrial RNA maturase gene transferred to the yeast nucleus can control mitochondrial mRNA splicing. Cell, 46, 837–844. - PubMed
-
- Barraille P., Chinestra,P., Bayard,F. and Faye,J.C. (1999) Alternative initiation of translation accounts for a 67/45 kDa dimorphism of the human estrogen receptor ERα. Biochem. Biophys. Res. Commun., 257, 84–88. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
